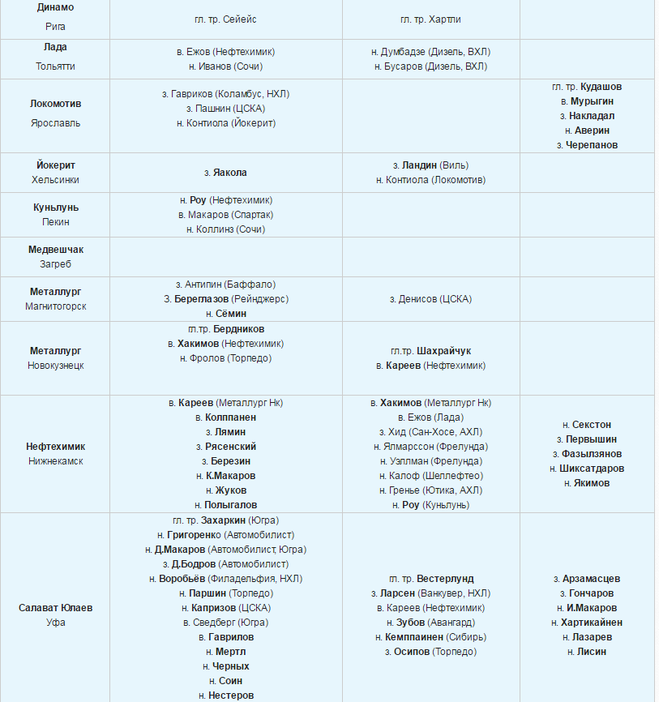
| гл. тренер. Владимир Воробьёв | ||
| защ. Никита Пивцакин («Спартак») | гл. тренер. Сергей Светлов | |
| вр. Евгений Аликин | нап. Андрей Алексеев («Динамо» Москва) | тренер. Павел Торгаев |
| защ. Михал Йордан | нап. Владимир Галузин («Нефтехимик») | тренер. Михаил Переяслов |
| защ. Глеб Корягин | нап. Георгий Меркулов (ХК «Сочи») | защ. Павел Турбин («Торпедо») |
| защ. Сергей Терещенко | нап. Дмитрий Константинов (ХК «Сочи») | нап. Денис Голубев («Сибирь») |
| нап. Владимир Михасёнок |
нап. Глеб Зырянов («Динамо» Рига) Глеб Зырянов («Динамо» Рига) |
нап. Кирилл Рассказов («Северсталь») |
| вр. Александр Евграфов | тренер. Александр Завьялов | защ. Никита Камалов (ХК «Сочи») |
| нап. Дмитрий Архипов | тренер. Игорь Петров | нап. Валентин Пьянов («Сибирь») |
| нап. Александр Шаров | нап. Андрей Локтионов (ЦСКА) | нап. Александр Полунин («Локомотив») |
| защ. Доминик Машин | вр. Дмитрий Лозебников (ЦСКА) | вр. Евгений Киселёв («Звезда», ВХЛ) |
| нап. Александр Горшков | нап. Илья Зубов («Спартак») | защ. Юрий Сергиенко |
| нап. Александр Кузнецов | нап. Радан Ленц («Белые Тигры», Чехия) Радан Ленц («Белые Тигры», Чехия) |
нап. Дмитрий Лугин |
| тренер. Олег Филимонов | нап. Давид Томашек («Спарта», Чехия) |
вр. Марек Лангхамер («Ильвес», Финляндия) |
| тренер. Станислав Басов | защ. Дмитрий Знахаренко («Динамо», Минск) | защ. Никита Зимин |
| тренер. Денис Богданов | вр. Патрик Бартошак («Пеликанс», Финляндия) | нап. Гинек Зогорна («Оскарсхамн», Швеция) |
| Остались | Пришли | Ушли | |
|
Динамо Мн |
в. з., Дмитрий Дерябин з., Илья Шинкевич з., Адам Альмквист (SWE) з., Павел Денисов з., Владислав Еременко н., Роман Горбунов (RUS) н., Игорь Мартынов н., Артем Демков н., Илья Усов н., Александр Когалев н., Денис Мосалев (RUS) н., Павел Варфоломеев (RUS) н., Кирилл Воронин (RUS) н., Владимир Алистров
н., Дмитрий Буйницкий |
в., Патрик Рыбар (SVK, «Кярпят», Финляндия) з., Лукас Бенгтссон (SWE, СКА, КХЛ) з., Сергей Сапего («Торонто Марлиз», AHL) з., Антон Линдхольм (SWE, «Рокфорд», АХЛ) з., Якуб Крейчик (CZE, «Йокерит», КХЛ) з., Дмитрий Коробов («Спартак»,
КХЛ) н., Мальте Стремвалль (SWE, СКА, КХЛ) н. н., Тейлор Бек (CAN, «Металлург» Мг, КХЛ) н., Александр Суворов («Динамо-Мл») н., Виталий Пинчук («Металлург») н., Сергей Кузнецов («Янгстаун», USHL) н., Тайлер Граовац (CAN, «Ванкувер», NHL)
н., Марио Кемпе (SWE, ЦСКА, КХЛ) |
в., Доминик Фурх (CZE, «Пльзень», Чехия) в., Дэнни Тэйлор в., Александр Осипков («Шахтер») в., Никита Толопило («Седертелье», Швеция Д2) з., Дмитрий Знахаренко («Амур», КХЛ) з., Владислав Колячонок («Сиракьюз», AHL) з., Степан Фальковский (СКА, КХЛ) з., Бреннан Менелл (USA, «Торонто», НХЛ) з., Илья Соловьев («Калгари», НХЛ) н., Вячеслав Грецкий («Неман») н. н., Алексей Протас («Херши», AHL) н., Брэндон Козун (CAN, «Амбри-Пиотта», Швейцария) н., Илья Литвинов («Академия Михайлова», ВХЛ) н., Франсис Паре («Авангард», КХЛ) н., Шейн Принс («Автомобилист», КХЛ) н., Райан Спунер (CAN, «Автомобилист», КХЛ) н., Николай Сытый («Торунь», ПХЛ) н., Артем Аносов («Лиепая», Латвия) н., Зак Митчелл (CAN, «Рапперсвиль», Швейцария) н., Андрей Павленко («Юность»)
н., Иван Дроздов («Спартак»,
КХЛ) |
| Пришли | Ушли | ||
|
Брест |
в. в., Андрей Ничипорчик з., Виталий Кафеев (RUS) з., Антон Киречко з., Роман Петручик н., Илья Балаболкин (RUS) н., Виктор Бовбель н., Денис Донской (RUS) н., Федор Качан н., Владислав Хадарович н., Андрей Колосов н., Ренат Сафин н., Дмитрий Самсонов (RUS) н., Алексей Селивоник н., Михаил Волошин н., Валерий Жигалов |
в., Иван Поляков («Неман») з., Виталий Марченко («Динамо-Мл») з., Данил Щеголев («Химик») з., Илья Сушко («Шахтер») з., Сергей Шелег (без клуба) з. н., Владимир Михайлов («Лида») н., Александр Пухловский («Пинские ястребы») н., Александр Семочкин («Локомотив»)
н., Никита Давыдов («Металлург-2») |
в., Степан Горячевских («Химик») з., Денис Грибко («Мёдон Кометс», Франция) з., Михаил Хоромандо («Неман») з., Владислав Микульчик («Шахтер») з., Камиль Мингазов (RUS, «Динамо-Алтай», ВХЛ-Б) з.,
Илья Бощук («Пинские ястребы») н., Сергей Пищук («Лида») н., Кирилл Батурин н., Антон Хомяков («Динамо-Мл») н., Валерий Кубраков
н. |
|
Остались |
Пришли | Ушли | |
|
Витебск |
в., Максим Журавский з., Илья Сидорук з., Максим Никитин з., Евгений Леонов з., Михаил Листопадов н., Максим Лемешевский н., Александр Гончаров н., Алексей Невидицин н., Роман Серафимович |
в., Никита Мытник («Авиатор») в., Кирилл Андреев («Юниор») з., Олег Станевич («Пинские ястребы») з., Даниил Мисюль («Юниор») з. з., Вадим Ерохо («Химик») з., Денис Кузьмин (RUS, «Южный Урал», ВХЛ) н., Владислав Ковалевич («Химик») н., Александр Левко («Шахтер») н., Егор Егоров («Могилев») н., Павел Мусиенко («Брест») н., Артур Гильманов (RUS, «Локомотив») н., Никита Ядроец («Нефтяник», ВХЛ) н., Магомед Гимбатов (RUS, СКА-Нева, ВХЛ) н., Никита Сошнин (RUS, «Барс», ВХЛ) н., Александр Щемель* («Динамо-Мл»)
н., Глеб Шкапцов* («Юниор») |
з., Кирилл Гусаров («Химик») з., Андрей Тимощенко («Юность») з., Кирилл Буйнич н., Никита Синельников («Металлург»)
н. н., Вадим Мурашов («Химик») н., Вадим Александрович н., Сергей Карпович («Юность») н., Кирилл Гриневич н., Сергей Адашкевич н., Илья Денисов н., Максим Ковальский |
|
Остались |
Пришли |
Ушли | |
|
Гомель |
в., Андрей Грищенко в., Максим Лубский з., Дмитрий Иванчиков з., Михаил Ковалев з., Максим Магалецкий з., Павел Воронов з., Вадим Сушко н., Кирилл Бовин н., Илья Камбович н., Евгений Хузеев н., Кирилл Куцырь н., Кирилл Межейников н. н., Иван Секерин
н., Николай Сусло |
з., Михаил Чурляев (RUS, «Лада», ВХЛ) з., Никита Устиненко (без клуба) з., Егор Юзленко («Чайка», МХЛ) з., Никита Стасенко (Беларусь U18) н., Евгений Дадонов («Шахтер») н., Игорь Карабанов («Шахтер») н., Станислав Кучкин (RUS, «Динамо-Мл») н., Павел Развадовский («Динамо-Мл») н., Илья Жуковский («Брест»)
н., Евгений Григоренко (RUS, «Лада», ВХЛ) |
з., Егор Иванов («Юность») з., Олег Попов (RUS, «Динамо» СПб, ВХЛ) з., Роман Рачинский (RUS, «Академия Михайлова», ВХЛ) з. н., Олег Качеловский («Барс», ВХЛ)
н., Нассер Субхи («Барс»,
ВХЛ) н., Булат Хамматов (KAZ) н., Герман Нестеров («Витязь», КХЛ) н., Александр Скоренов (СКА-Нева, ВХЛ) н., Павел Печур («Могилев») н., Антон Елисеенко («Сокол», УХЛ)
н., Максим Войтович («Нефтехимик»,
КХЛ) |
|
Остались |
Пришли |
Ушли | |
|
Динамо-Мл |
з., Богдан Зозон з., Антон Седов з., Геннадий Косинский н., Павел Копытин (RUS) н., Антон Мохорев н., Никита Пышкайло
н. |
в., Михаил Карнаухов («Металлург») в., Ян Шостак («Минские зубры») в., Павел Капский («Витебск») з., Илья Летов («Металлург») з., Михаил Калач («Соболь») з., Максим Глинский (Беларусь U18) з., Владислав Сурков («Минские зубры») з., Даниил Старинский («Минские зубры») з., Дмитрий Клещик («Минские зубры») н., Эдуард Гринь («Краматорск», УХЛ) н., Александр Каракулько («Днепр», УХЛ) н., Максим Слыш («Могилев») н., Леонид Апанасюк («Минские зубры») н., Мирослав Михалев (Беларусь U18) н., Иван Аношко (Беларусь U18) н., Владимир Онуфриюк («Минские зубры») н., Дмитрий Поцелуйко («Минские зубры») н., Кирилл Игнатов («Металлург») н., Матвей Курилович («Минские зубры»)
н. |
в., Алексей Колосов («Динамо» Мн, КХЛ) в., Ян Шелепнев в., Никита Толопило («Седертелье», Швеция Д2) з., Павел Денисов («Динамо» Мн, КХЛ) з., Андрей Геращенко («Ижсталь», ВХЛ) з., Тимофей Ковгореня («Спокейн Чифс», WHL) з., Дмитрий Кузьмин («Виннипег», НХЛ) з., Илья Соловьев («Калгари», НХЛ) з., Виталий Марченко («Брест») з., Александр Шкрабов («Торунь», ПХЛ) н., Николай Сытый («Торунь», ПХЛ) н., Владимир Алистров («Динамо» Мн, КХЛ) н., Дмитрий Амброжейчик («Шахтер») н., Артем Аносов (ХК «Лиепая», Латвия) н., Вячеслав Грецкий («Неман») н., Егор Клавдиев («Летбридж», WHL) н., Илья Литвинов («Академия Михайлова», ВХЛ) н., Игорь Мартынов («Динамо» Мн, КХЛ) н., Кирилл Никулин («Шахтер») н., Андрей Павленко («Юность») н., Павел Развадовский («Гомель») н. н., Артем Волков (завершил карьеру)
н., Александр Щемель («Витебск») |
|
Остались |
Пришли |
Ушли |
|
|
Лида |
в., Павел Шелесный з., Никита Кардашев з., Денис Малишевский з., Кирилл Тарасевич з., Никита Волес з., Виктор Вусик н., Денис Зайчик н., Никита Бастрыкин н., Владислав Серяков н., Александр Бунец н., Вадим Апанасевич н., Валерий Будевич н. н., Егор Шкляревский
н., Александр Идиатуллин |
в., Константин Козловский («Локомотив») з., Александр Кудрявцев 1992 («Пинские ястребы») з., Даниил Рогач (Беларусь U18) з., Алексей Субоч («Неман-2») з., Владислав Мартынюк (без клуба) з., Николай Козлов (RUS, СКА-Нева, ВХЛ) з., Никита Зубов* (без клуба) н., Илья Казьянин («Металлург») н., Сергей Пищук («Брест») н., Антон Иванковский («Гомель») н., Роман Романчук («Неман-2») н., Даниил Собко (без клуба) н., Владимир Михайлаки (RUS, ХК «Белгород», НМХЛ) н., Владислав Ануфриенко* («Брест-2»)
н. |
в., Виктор Мойсеенко («Неман») в., Роман Долголевец з., Егор Кудин з., Андрей Гостев («Локомотив») з., Олег Сиренко (RUS) н., Владимир Михайлов («Брест») н., Никита Бастрыкин («Неман») н., Андрей Перегуда («Локомотив») н., Павел Марщенок (завершил карьеру) н., Кирилл Рашевский н., Алексей Бусько н., Ярослав Смычков («Бобруйск-2») н., Егор Гайдук («Бобруйск-2»)
н., Валерий Будевич («Неман-2») |
|
Остались |
Пришли |
Ушли |
|
|
Локомотив |
в. з., Владислав Гончаров з., Павел Курдюков (RUS) з., Андрей Филичкин н., Илья Цулыгин н., Руслан Гайсин (RUS) н., Павел Дашков н., Дмитрий Колышкин
н., Максим Мельников (RUS) |
в., Сергей Степанов («Шахтер») з., Андрей Гостев («Лида») з., Илья Рак («Неман») з., Дмитрий Буровцев (без клуба) з., Виталий Тесленко (RUS, «Буран», ВХЛ) з., Антон Федоренчик («Юниор») з., Денис Черевач («Металлург») н., Артур Абметка («Неман») н., Максим Субботин (RUS, «Могилев») н., Андрей Перегуда («Лида») н., Евгений Кунцевич (без клуба) н., Арсений Спивак («Бобруйск») н., Виктор Масилевич («Авиатор») н., Иван Некрасов («Шахтер-2») н., Роман Еремейчик («Минские зубры») н. н., Федор Пышкайло («Пинские ястребы»)
н., Александр Чабан («Бобруйск-2») |
в., Константин Козловский («Лида») з., Михаил Прус («Витебск») з., Владислав Барковский («Юность») з., Олег Горошко («Металлург») з., Андрей Шикуть («Могилев») з., Никита Тарлецкий («Могилев») н., Егор Гайнетдинов (RUS, «Юность») н., Артур Гильманов (RUS, «Витебск») н., Артемий Черников («Кулагер», Казахстан) н., Александр Семочкин («Брест») н., Вячеслав Шарипов (RUS, «Химик») н., Артур Савинов («Неман») н., Владислав Рядченко («Шахтер»)
н. |
|
Остались |
Пришли |
Ушли |
|
|
Металлург |
з., Артем Волченков з., Владимир Ерменков н., Василий Медведев н., Юрий Веремеенко н., Евгений Соломонов н., Алексей Михнов (RUS) н., Захар Полищук н., Дмитрий Войтехов н., Илья Спат н., Егор Воронов н., Максим Чубуков |
гл. тр., Дмитрий Кравченко в., Роман Бобарико («Пинские ястребы») в., Александр Самойлов (RUS, «Звезда», ВХЛ) з. з., Олег Горошко («Локомотив») з., Владислав Габрусь («Могилев») з., Павел Купчихин (RUS, «Ирбис», МХЛ) з., Данил Мокрушев (RUS, «Челмет», ВХЛ) з., Вячеслав Грамович (Беларусь U18) з., Владислав Наумов (Беларусь U17) н., Георгий Кришталь («Юниор») н., Никита Синельников («Витебск») н., Андрей Лошко (Беларусь U18) н., Илья Морозов (Беларусь U18) н., Александр Тимирев (RUS, «Дизель», ВХЛ) н., Артем Кислый («Шахтер») н., Вячеслав Андрющенко («Южный Урал», ВХЛ) н., Илья Шуринов («Чайка», МХЛ) н., Артем Сафонов (Беларусь U17)
н., Алексей Мастрюков (RUS, «Лада», ВХЛ) |
гл. в., Дмитрий Мильчаков (Академия Михайлова, ВХЛ) в., Михаил Карнаухов («Динамо-Мл») з., Денис Черевач («Локомотив») з., Сергей Копылец («Дунареа», Румыния) з., Илья Летов («Динамо-Мл») з., Владислав Соколовский («Донбасс», УХЛ) з., Дмитрий Заламай («Шахтер») з., Александр Еронов («Шахтер») н., Евгений Астанков («Металлург» Нк, ВХЛ) н., Алексей Ефименко (завершил карьеру) н., Руслан Журня («Химик») н., Виталий Пинчук («Динамо» Мн, КХЛ) н., Василий Филяев («Автомобилист», КХЛ) н., Антон Угольников (RUS, «Академия Михайлова», ВХЛ) н., Илья Казьянин («Лида») н., Кирилл Игнатов («Динамо-Мл»)
н. |
|
Остались |
Пришли |
Ушли |
|
|
Могилев |
в., Владислав Карачун з., Ярослав Маслеников з., Максим Прохоренко з., Егор Долкарт з., Егор Калин з., Александр Алешков н., Максим Попков н., Владислав Шутов н., Данила Гапеев н., Иван Коташенко н., Александр Козинов н., Артем Васько н., Виталий Вальков н., Илья Шапиро н., Матвей Винкевич
н., Илья Мищенко |
гл. тр., Александр Матерухин в., Артур Малков (RUS, «Пинские ястребы») з. з., Андрей Клепча («Пинские ястребы») з., Олег Качесов (RUS, АКМ, МХЛ) з., Юрий Жуковский («Юниор») з., Андрей Шикуть* («Локомотив») з., Никита Тарлецкий* («Локомотив») н., Глеб Былина («Пинские ястребы») н., Александр Попков («Авиатор») н., Артем Шапиро («Авиатор») н., Дмитрий Корелов («Авиатор») н., Павел Печур («Гомель») н., Владислав Сайко (KAZ, «Снежный барс», Казахстан) н., Герман Поддубный (RUS, «Сарыарка», Казахстан) н., Ярослав Билищук («Химик»)
н., Николай Мирончик (без
клуба) |
гл. тр., Дмитрий Рыльков в., Артем Сергеев (RUS) з., Алексей Костерев («Химик») з. з., Андрей Герасимов (RUS) з., Владислав Мирошкин (завершил карьеру) з., Дмитрий Данилюк н., Владислав Мисников (RUS) н., Максим Слыш («Динамо-Мл») н., Виктор Андрущенко («Сокол», УХЛ) н., Егор Егоров («Витебск») н., Александр Матерухин (завершил карьеру) н., Сергей Кукушкин (завершил карьеру) н., Игорь Лапшов (RUS, «Динамо» СПб, ВХЛ)
н., Максим Субботин (RUS, «Локомотив») |
|
Остались |
Пришли |
Ушли |
|
|
Неман |
в. з., Никита Крыскин з., Сергей Романович н., Павел Ажгирей н., Егор Степанов н., Игорь Варивончик н., Артур Буйницкий н., Алексей Фурса н., Игорь Козячий н., Артем Левша н., Александр Малявко н., Сергей Малявко н., Виталий Зацепилин (RUS) н., Павел Пищальников (RUS)
н., Павел Зыков (RUS) |
в., Максим Малютин («Шахтер») в., Виктор Мойсеенко («Лида») з., Михаил Хоромандо («Брест») з., Дмитрий Костромитин (RUS, «Краковия», ПХЛ) з., Алексей Власкин (RUS, «Алматы», Казахстан) з., Антон Гришанов («Звезда», ВХЛ) н., Павел Боярчук (без клуба) н., Артур Савинов («Локомотив») н., Никита Ремезов («Шахтер»)
н. |
в., Виталий Трус («Краматорск», УХЛ) в., Иван Поляков («Брест») з., Эдуард Насыбуллин (RUS, «Буран», ВХЛ) з., Данила Паливко («Адмирал», КХЛ) з., Илья Рак («Локомотив») з., Артем Волков (RUS) з., Матвей Божко н., Артур Абметка («Локомотив») н., Кристиан Дзюбиньски (POL, «Уния», ПХЛ)
н., Дмитрий Каштанов (RUS, «Химик») |
|
Остались |
Пришли |
Ушли |
|
|
Химик |
в., Артем Шпилев (RUS)
в. з., Евгений Еременко (RUS) з., Максим Каменьков з., Никита Ермачков з., Алексей Садовик з., Александр Исаев з., Яков Усович з., Руслан Трубкин (RUS) з., Алексей Храповицкий н., Виктор Гавриленко н., Михаил Бучкин н., Максим Кривченя н., Артем Землянкин н., Артем Ментюк н., Артем Балтрук н., Денис Кирпиченок |
в., Степан Горячевских («Брест») з., Кирилл Гусаров («Витебск») н., Дмитрий Каштанов (RUS, «Неман») н., Вячеслав Шарипов (RUS, «Локомотив») н. н., Руслан Журня («Металлург») н., Вадим Мороз (Беларусь U18) н., Даниил Сотишвили (Беларусь U18) н., Егор Калугин («Витебск»)
н., Вадим Мурашов («Витебск») |
з., Георгий Пузатов (RUS) з., Вадим Ерохо («Витебск») з., Данил Щеголев (RUS, «Брест») н., Игорь Коваленя (завершил карьеру) н., Ярослав Билищук («Могилев») н., Константин Пименов (RUS) н., Владислав Ковалевич («Витебск») н., Евгений Фарафонтов («Шахтер») н., Игорь Леоненко (завершил карьеру)
н., Владислав Лоновенко |
|
Остались |
Пришли |
Ушли |
|
|
Шахтер |
з. з., Олег Пожиган з., Сергей Станкевич з., Максим Толопило з., Алексей Гарапучик н., Виталий Антонович н., Денис Белоусов н., Богдан Хамицевич н., Роман Малиновский н., Александр Жидких
н., Андрей Феклистов |
в., Игорь Брикун («Подхале», ПХЛ) в., Александр Осипков («Динамо» Мн, КХЛ) з., Владислав Микульчик («Брест») з., Арсений Сазанович (Беларусь U18) з., Дмитрий Заламай («Металлург») з., Александр Еронов («Металлург») з., Даниил Бокун («Динамо» СПб, ВХЛ) з., Роман Дюков («Химик», ВХЛ) н., Дмитрий Амброжейчик («Динамо-Мл») н., Евгений Фарафонтов («Химик») н., Алексей Котеловский («Пинские ястребы») н., Кирилл Никулин («Динамо-Мл») н. н., Владислав Рядченко («Локомотив») н., Дмитрий Пестунов (RUS, «Дебрецен», Венгрия)
н., Павел Чернов (RUS, «Спартак», КХЛ) |
в., Максим Малютин («Неман») в., Сергей Степанов («Локомотив») з., Сергей Боголейша («Ястшембе», ПХЛ) з., Арсений Борисов («Торпедо», Казахстан) з., Роман Достанко («Юность») з., Максим Сергеев (RUS, «Рязань», ВХЛ) з., Артем Лесников (RUS, «Юность») з., Илья Сушко («Брест») н., Данила Карабань («Арлан», Казахстан) н., Роман Крикуненко (RUS, «Югра», ВХЛ) н., Игорь Карабанов («Гомель») н., Евгений Дадонов («Гомель») н., Артем Кислый («Металлург») н., Никита Ремезов («Неман») н. н., Владимир Джиг («Дизель», ВХЛ) н., Егор Иванов (RUS, «Южный Урал», ВХЛ)
н., Александр Левко («Витебск») |
|
Остались |
Пришли |
Ушли |
|
|
Юность |
в., Сергей Большаков (RUS) в., Ростислав Зиновенко з., Андрей Антонов з., Никита Парфенюк з., Вадим Щечин з., Алексей Виноградов (RUS) н., Кирилл Брикун н., Сергей Дрозд н., Всеволод Кашкар н., Александр Пальчик
н. |
з., Владислав Барковский («Локомотив») з., Роман Достанко («Шахтер») з., Егор Иванов («Гомель») з., Андрей Тимощенко («Витебск») з., Максим Сущинский (Беларусь U18) з., Артем Лесников* (RUS, «Шахтер») н., Дмитрий Арсенюк (RUS, «Дизель», ВХЛ) н., Сергей Демагин («Арлан», Казахстан) н., Егор Дугин (RUS, «Куньлунь», КХЛ) н., Иван Фищенко (RUS, «Югра», ВХЛ) н., Егор Гайнетдинов (RUS, «Локомотив») н., Владимир Ровба («Пинские ястребы») н., Андрей Павленко («Динамо» Мн, КХЛ) н., Федор Николаеня (Беларусь U18)
н., Владислав Шило
(Беларусь U18) |
в., Александр Осипков («Шахтер») з. з., Павел Казакевич (завершил карьеру) з., Никита Мицкевич («Заглембе», ПХЛ) з., Евгений Ногачев («Донбасс», УХЛ) з., Дмитрий Шуленин (RUS, «Торпедо», Казахстан) з., Никита Сиротин («Лада», ВХЛ) н., Егор Буяльский («Челмет»,
ВХЛ) н., Валентин Демченко («Спартак», КХЛ) н., Даниил Мартынов н., Александр Когалев («Динамо» Мн, КХЛ) н., Евгений Ковыршин (завершил карьеру) н., Георгий Кришталь («Металлург») н., Станислав Лопачук («Амьен», Франция) н., Денис Мингалеев (RUS, «Южный Урал», ВХЛ) н., Евгений Оксентюк (Северная Америка) н., Максим Рассейкин (RUS, «Автомобилист», КХЛ) н., Михаил Стефанович («Донбасс», УХЛ) н., Кирилл Тютяев (RUS, «Гранд Рэпид», AHL)
н. |
 , Роб
Клинкхаммер (CAN, «Динамо»
Мск, КХЛ)
, Роб
Клинкхаммер (CAN, «Динамо»
Мск, КХЛ) , Сергей Бронников (RUS)
, Сергей Бронников (RUS)
 , Кирилл
Куприянчик («Минские зубры»)
, Кирилл
Куприянчик («Минские зубры») , Павел Мусиенко («Витебск»)
, Павел Мусиенко («Витебск»)
 , Михаил
Прус («Локомотив»)
, Михаил
Прус («Локомотив») , Егор Калугин («Химик»)
, Егор Калугин («Химик»)
 , Игорь Ревенко
, Игорь Ревенко , Андрей
Алексеев (RUS, «Сокол»,
УХЛ)
, Андрей
Алексеев (RUS, «Сокол»,
УХЛ) , Даниил Степанов
, Даниил Степанов , Егор Игнатенко (Беларусь
U18)
, Егор Игнатенко (Беларусь
U18) , Александр
Суворов («Динамо» Мн, КХЛ)
, Александр
Суворов («Динамо» Мн, КХЛ) , Игорь Спешилов
, Игорь Спешилов , Вячеслав Аксенов* («Витебск-2»)
, Вячеслав Аксенов* («Витебск-2») , Алексей Мерзлов
, Алексей Мерзлов , Алексей
Черников («Шахтер-2»)
, Алексей
Черников («Шахтер-2») , Тигран Манукян (RUS)
, Тигран Манукян (RUS) , Алексей
Бадун (без клуба)
, Алексей
Бадун (без клуба) тр., Анатолий Степанищев
тр., Анатолий Степанищев , Сергей Тополь (RUS)
, Сергей Тополь (RUS) , Павел
Голубич («Сокол», УХЛ)
, Павел
Голубич («Сокол», УХЛ) , Владислав
Габрусь («Металлург»)
, Владислав
Габрусь («Металлург») , Максим Городецкий
, Максим Городецкий , Вячеслав Грецкий («Динамо»
Мн, КХЛ)
, Вячеслав Грецкий («Динамо»
Мн, КХЛ) , Вадим Вашкевич
, Вадим Вашкевич
 , Артем
Логунов («Пинские ястребы»)
, Артем
Логунов («Пинские ястребы»)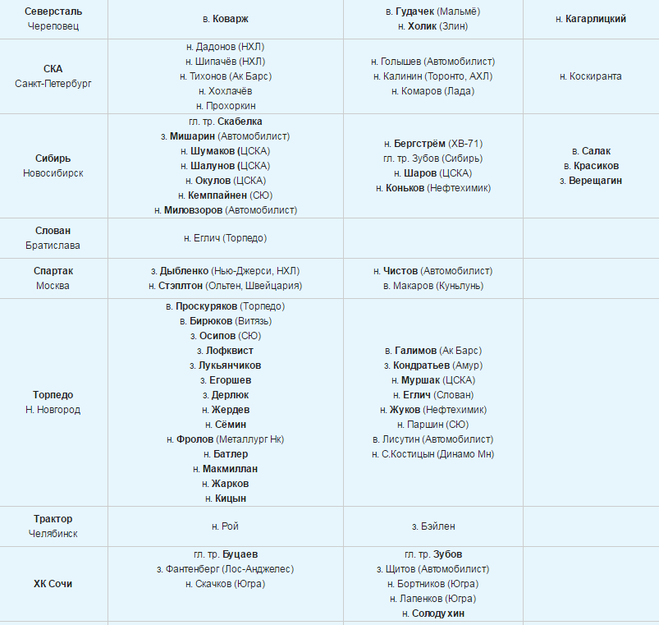 , Илья Казнадей
, Илья Казнадей , Валентин Разумняк
(RUS, «Химик», ВХЛ)
, Валентин Разумняк
(RUS, «Химик», ВХЛ) , Валерий Бочкарев (RUS, «Лада», ВХЛ)
, Валерий Бочкарев (RUS, «Лада», ВХЛ) , Александр Китаров
, Александр Китаров , Кирилл
Дьяков (RUS, «Сочи»,
КХЛ)
, Кирилл
Дьяков (RUS, «Сочи»,
КХЛ) , Максим Парфеевец (завершил
карьеру)
, Максим Парфеевец (завершил
карьеру)ВХЛ. Изменение составов команд. Межсезонье-19 — Колумнисты
сводка о переходах игроков и тренеров в ВХЛ
Стандартный шрифт обозначает состоявшийся переход;
курсивом обозначен пробный контракт;
добавление символа * — неподтверждённая информация
«Барс» Казань (фарм-клуб «Ак Барса»)
тренерский штаб: гт.Душкин («Зауралье») т.Э.Горбачёв («Ирбис») т.Балмин т.Агопеев
——————
покинули тренерский штаб: гт.Кадейкин т.А-м Анисимов (оба в «Ирбис»)
игроки остались: в.А.Мифтахов в.А.Мисбахов з.Богатов з.Е.Федин з.К.Лучевников з.Д.Мужиков з.Цуканов з.Марин з.Ляхов н.Редков н.Сошнин н.Т.Елисеев н.А-м Валеев н.Пестушко н.М.Марушев н.Ил.Сафонов н.М.Лазарев н.Емец н.И.Хасанов
——————
новые игроки: н.Вильданов («Ирбис») н.Левитский н.Д.Лапин (оба из «Ермака») з.Г.Мальцев («Тайфун») з. О.Железнов н.Хапов в.И.Голубев з.Адамчук н.Д.Воеводин (все из «Нефтяника», через АБ) н.Кара в.Э.Гарипов (оба из «Ак Барса»)
О.Железнов н.Хапов в.И.Голубев з.Адамчук н.Д.Воеводин (все из «Нефтяника», через АБ) н.Кара в.Э.Гарипов (оба из «Ак Барса»)
——————
игроки выбыли: з.Э.Насыбуллин («ORG») н.Тимиров (ЦСК ВВС, аренда) з.Д.Журавлёв («Ак Барс») н.К.Зиновьев («Алматы») з.Мих.Сидоров («Буран», через «Локомотив») з.Т.Фаткуллин («Нефтяник», в аренду) н.Фазлеев («Нефтехимик», через «Ак Барс») з.Д.Курашов («Темиртау») н.Безносов («Красноярские Рыси») н.М-т Ибрагимов н.Шинкаренко н.Д.Литвинов з.М-м А Орлов
«Буран» Воронеж (фарм-соглашение с «Локомотивом»)
тренерский штаб: гт.Ярушкин (юн.сб.России-2002гр) ст.Д.Фролов (экс-«Химик») мдж/т.М.Бирюков
——————
покинули тренерский штаб: гт.А.Трофимов («Крылья Советов») т.Вит.Вл.Карамнов т.Ал-р Л Данилов
игроки остались: з.Дм.А.Кузнецов н.Алёшин н.Шипилов н.С-а Андреев з.М.Сергеев н.Акиньшин
——————
новые игроки: з.Вад.Ефимов («Россошь») н.П.Антипов з.П.Лукошин (оба из «Кристалла» С) н. Д-а Цыганов («Нефтяник») з.Н.Громов («Локо», скрытая аренда) з.Г.Штайгер з.Ал-р О Калинин з.Ант.Малышев н.В.Рыбаков (все из «Локо») з.Ю.Маслов («Зауралье») н.Чиглинцев н.А.Князев-ст (оба из «Рубина») в.Аляпкин («Лида») з.Цирулёв («Алматы») н.Шацкий («Рязань») з.Рад.Ахметгалиев («Ирбис») н.Кляузов («Химик») з.И.Ковалёв-аренда н.Червоненко н.Мих.Беляев н.Аноховский-аренда в.Д.Исаев н.Слепец (все из «Локомотива») н.Ив.П.Козлов («Валь-д’Ор», QMHL через аренду от «Локомотива») в.Андр.О.Макаров (аренда от «Автомобилиста») н.И.Романов («Лада», через «Локомотив») з.Мих.Сидоров («Барс», через «Локомотив») в.Анаркулов («Молот»)
Д-а Цыганов («Нефтяник») з.Н.Громов («Локо», скрытая аренда) з.Г.Штайгер з.Ал-р О Калинин з.Ант.Малышев н.В.Рыбаков (все из «Локо») з.Ю.Маслов («Зауралье») н.Чиглинцев н.А.Князев-ст (оба из «Рубина») в.Аляпкин («Лида») з.Цирулёв («Алматы») н.Шацкий («Рязань») з.Рад.Ахметгалиев («Ирбис») н.Кляузов («Химик») з.И.Ковалёв-аренда н.Червоненко н.Мих.Беляев н.Аноховский-аренда в.Д.Исаев н.Слепец (все из «Локомотива») н.Ив.П.Козлов («Валь-д’Ор», QMHL через аренду от «Локомотива») в.Андр.О.Макаров (аренда от «Автомобилиста») н.И.Романов («Лада», через «Локомотив») з.Мих.Сидоров («Барс», через «Локомотив») в.Анаркулов («Молот»)
——————
игроки выбыли: в.Г.Кузнецов («Динамо» Мск) з.Ег.Ю.Орлов з.Казаманов з.Толпегин н.Бицадзе (все в «Динамо» Тв, через «Динамо» Мск) н.Бешаров («Динамо» Тв) з.А.Смолин н.Хасаншин (оба в «Южный Урал») н.А.Буйницкий (завершил игровую карьеру) з.Слепцов («Тамбов») в.Хомутов («Иртыш») з.С.Алексеев («Химик») н.Д-л Попов («Россошь») н.Я.Хоменко («Алтай-Торпедо») н. Пушкарский («Сахалин») н.Мурзин («Южный Урал») з.Беленький н.И.Мирнов н.Анд.Ан.Белозёров в.Ал-р Зубарев н.Черепуха з.Суховерхов з.А.Прокопенко н.Чернявский в.Корняков
Пушкарский («Сахалин») н.Мурзин («Южный Урал») з.Беленький н.И.Мирнов н.Анд.Ан.Белозёров в.Ал-р Зубарев н.Черепуха з.Суховерхов з.А.Прокопенко н.Чернявский в.Корняков
«Горняк» Учалы (фарм-клуб «Автомобилиста»)
тренерский штаб: гт.О.Леонтьев т.Симаков (дебют) т.Ганеев сп.дир.Айр.Нургалиев
——————
покинули тренерский штаб: ст.С.Могильников («Иртыш»)
игроки остались: з.Корчемкин з.И.Сушинский з.Галюк з.З.Богатырёв з.Переляев з.Ал-р Севостьянов н.Вяч.Шарипов н.Митякин н.Ивашов н.Мих.Назаров н.Мингалеев н.Еркин н.Белоцкий н.Д.Носков н.П.Воронков н.Протапович
——————
новые игроки: з.В.Леонтьев («Ладья») в.В.Галкин н.Дербенёв (оба из «Авто») н.Гайдеик («Мамонты Югры») н.Грымзин з.Катин (оба из «Югры») в.Старостин («Зауралье») з.С.Козлов («Рубин») н.Д.Жукенов («Металлург» Нк) н.А.Юшков («Ермак», через аренду от «Ак Барса») н.В.Кузнецов («Автомобилист») з.Галыгин («Крылья Советов», через АЕ) з.Ф.Беляков («Звезда», через АЕ) н.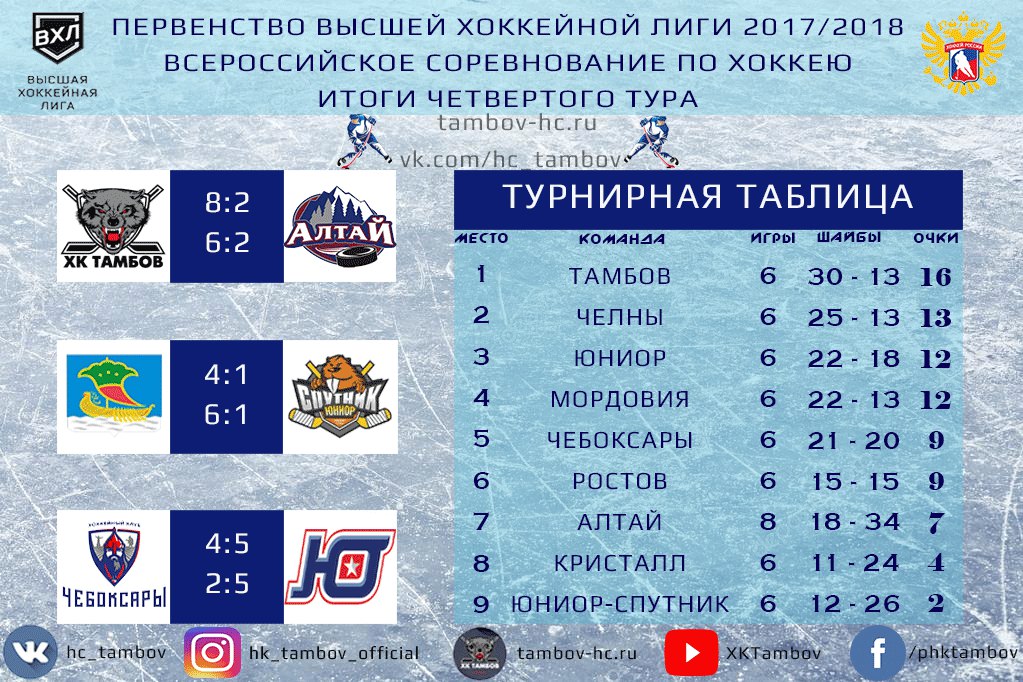 М.Бякин («Звезда»)
М.Бякин («Звезда»)
——————
игроки выбыли: з.Аблаев («Нефтяник») з.Арс.Нургалиев н.Д.Антонов (оба в «Южный Урал») н.Ревацкий («Тамбов») н.Арсенюк н.Борисенков (оба в «Югру») н.Ант.Кошелев («Темиртау») н.С.Третьяков («Рязань») з.Ю.Журавлёв з.Мирочников н.М-м А Седов (все в «Авто») в.Гайдуллин («Ценг Тоу») в.Р.Голованов («Хумо») з.Цапаев («Сахалин») з.Дубовик («Номад») н.Н.Сафронов н.Русл.Сафин з.Лебединец з.Юзеев
«Дизель» Пенза
тренерский штаб: гт.А.Ваулин т.Шандуров т.Ан.Агапов т.Ал.Шакиров (дебют) гм.Лопушанский («Нефтяник-2003»)
——————
покинули тренерский штаб: гм.Артёмкин
игроки остались: в.М.Борисов з.Люлько з.Дм.Киселёв з.Забавин з.Д.Бельский н.Думбадзе н.Н.Дёмин н.Колмыков н.Д-а Попов н.В.Барулин н.Дан.Макаров
——————
новые игроки: з.Вл-р Марченко («Тамбов») н.Байкеев («Рубин») н.Андриянов н.С.В.Абрамов (оба из «Молота») в.Гафиуллин («Металлург» Нк, аренда) н.Мокшанцев («Гап») н.Н.Ли («Торос») з. З.Подзиньш («Алматы») н.Пав.В.Медведев («Металлург» Нк) з.М.Мацейко («Ницца») н.Тврдонь («Краковия») з.П.Володин («Зауралье») в.О.Д.Назаров («Ермак») н.В.Хабаров («Ижсталь») з.С.Романов («Витязь»)
З.Подзиньш («Алматы») н.Пав.В.Медведев («Металлург» Нк) з.М.Мацейко («Ницца») н.Тврдонь («Краковия») з.П.Володин («Зауралье») в.О.Д.Назаров («Ермак») н.В.Хабаров («Ижсталь») з.С.Романов («Витязь»)
——————
игроки выбыли: н.Вл-в Волков («Рязань») в.Фокин («Сахалин») в.Носов («Зауралье», через «Металлург» Мг) з.Е.Артамонов з.Семичастнов (оба в «Зауралье») з.Н.Зеленин («Молот») з.Корабейников н.Зюзякин («Химик») н.Ю.Кокшаров («Гомель») н.В.Москалёв («Сарыарка») н.Кокшин («Чебоксары») н.Ал-р Тарасов («Торпедо» У-К) н.Сулев («Красная Армия») н.А.Дудин н.Чумаков н.Кантеев (все в «Юниор») з.Анд.Соколов («Кузнецкие Медведи») н.Савунов («СКА-Нева», через СКА) з.А.Антонов («Ростов») н.С.Колесников («Авто»)
«Динамо» Санкт-Петербург (фарм-соглашение с «Витязем»)
тренерский штаб: гт.Э.Занковец (экс-«Слован») т.Занатта т.С.Громов («Динамо-2008», Минск) т.П.Черкас т.В.Занковец («Ак Барс»)
——————
покинули тренерский штаб: гт.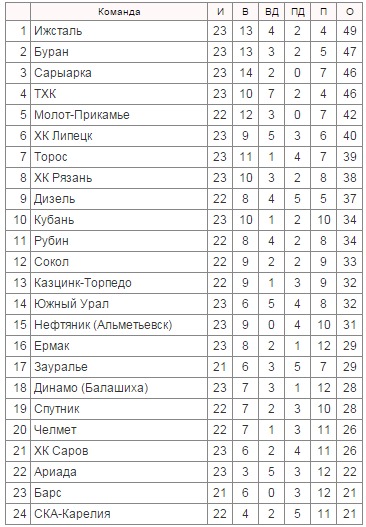 Башкатов т.А-м Караваев (оба в МХК «Динамо СПб»)
Башкатов т.А-м Караваев (оба в МХК «Динамо СПб»)
игроки остались: в.Тодыков з.Ручкин з.Кремзер з.Вен.Баранов з.Буханко н.С.Кондратьев н.Дан.Жарков н.А.Нечаев н.А-м Ал Чернов н.Дыняк н.М.Джиошвили н.Пухов н.П.Ротенберг н.А.Назаревич н.В.Барабанов н.А.Кулагин н.Жабреев з.Власкин
——————
новые игроки: н.Кр.Афанасьев з.Васин з.Сулима (все из МХК «Динамо СПб») з.Кожемякин з.Паутов н.Н.Гончаров в.Сапрыкин (все из «Витязя») в.Синягин з.И.Гавриленко (оба из «Южного Урала») з.Ушнев («Ценг Тоу») н.Гаврус («Динамо-Молодечно») н.Рычагов («Югра») з.Кудинов (возобновил игровую карьеру) н.В.Зорин («Орлик»)
——————
игроки выбыли: н.Монс н.И.Петраков (оба в «Витязь») н.Дм.Уваров н.Пластинин (оба в «Динамо» Тв) н.Шипов н.Верба н.Ант.Васильев (все в «Динамо» Тв, через «Динамо» Мск) н.С.Шестаков з.Молодцов (оба в «Торпедо» У-К) з.Назаркин («Челмет», через «Трактор») н.А.Денежкин («Лада») з.Ив.А.Воробьёв н.К.Кузнецов з.П.Лукин н.Анд.Бирюков (все в «Сарыарку») н.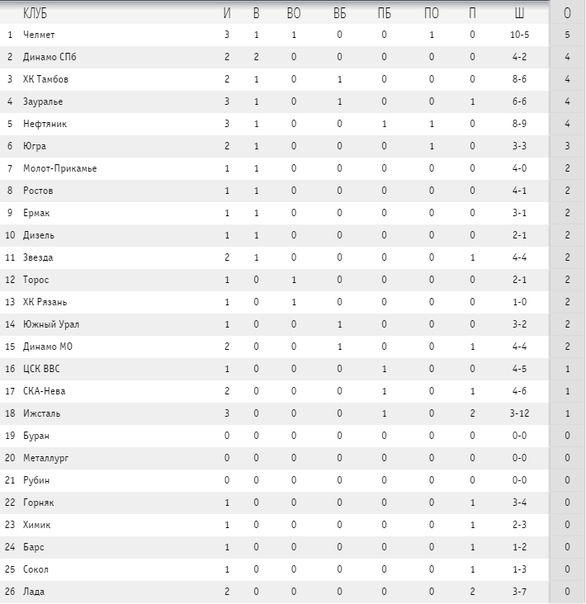 Жихарев («Ермак») в.Янин (завершил игровую карьеру) з.Дм.Ермошенко
Жихарев («Ермак») в.Янин (завершил игровую карьеру) з.Дм.Ермошенко
«Динамо» Тверь (фарм-клуб «Динамо» Мск)
тренерский штаб: гт.Орешкин (экс-«Динамо» СПб) ст.А.Антипов т.Дм.М.Попов (оба — экс-МХК «Динамо» Мск) т.А.Заболотнев («Атланты») т.Сыченко («Металлург» Нк) т.Зеленов (МХК «Динамо» Мск)
игроки: з.Ег.Ю.Орлов з.Казаманов з.Толпегин н.Бицадзе (все из «Бурана», через ДМо) н.Бешаров («Буран») з.Грищенко н.Аль-Джилауи н.Чувилов з.Семенеко (все из МХК «Динамо» Мск) н.П.Ткаченко («Сибирь», через ДМо) н.С.Панкратов н.Ал.Петрунин (оба из «Динамо» Мск) в.Тяло н.Ег.А.Попов (оба из «Рубина», через ДМо) з.Озеров (экс-МХК «Динамо» Мск, через ДМо) н.Гусынин (экс-«Снежные Барсы», через ДМо) з.Кармашков («Адмирал», через ДМо) з.Грязев («Торос») н.Дм.Уваров н.Пластинин (оба из «Динамо» СПб) н.Шипов н.Верба н.Ант.Васильев (все из «Динамо» СПб, через ДМо) н.Ив.Анд.Иванов («Рязань») н.Авдюхин («DME Hockey Academy», USPHL) н.О.Зайцев («Ред Дир», WHL) н.Бусаров («С. Юлаев», через ДМо) з.Кобелев («Химик») з.А.Осин («Дизелист») з.Щенков («Ижсталь») з.С.Степанов («Орлик»)
Юлаев», через ДМо) з.Кобелев («Химик») з.А.Осин («Дизелист») з.Щенков («Ижсталь») з.С.Степанов («Орлик»)
«Ермак» Ангарск (фарм-соглашение с «Адмиралом»)
тренерский штаб: ио.гт.Д.Крамаренко (экс-тренер) т.А.Потапов мдж.С.Гордеев
——————
покинули тренерский штаб: гт.О.Болякин («Алтай» У-К)
игроки остались: в.В.Никитин в.Дорожко з.Ал-й Ложкин з.А.Шестопалов н.А-м Воробьёв н.Раенко н.Чубыкин н.Трапезников н.В.Бочкарёв н.А-м Севостьянов н.С.Станкевич з.С.Сухов (аренда от «Сибири»)
——————
новые игроки: з.Коренков («Зауралье») з.А.Краснов («Звезда») н.Мельник н.Каргин (оба из «Ермак-ЮХЛ») н.Градусов з.Ельшанский н.Тымченко н.Г.Сергеенко з.Шикун (все из «Молота») н.П.Горохов («Русские Витязи») з.Е.Дубовицкий («Ценг Тоу») н.Жихарев («Динамо» СПб) з.Ахияров («Толпар», через «Адмирал») н.Дельнов («Молот», через «Адмирал») н.К.Кожевников («Югра», через «Адмирал») н.Казаковцев («Северсталь») н.Мишкин («Крылья Советов») н. А-м Осипов («Нефтяник»)
А-м Осипов («Нефтяник»)
——————
игроки выбыли: н.Н.Коротков («Сибирь», возврат аренды) н.Левитский н.Д.Лапин (оба в «Барс») з.Анд.Конев («Сахалин») з.Р.Осипов («Рязань») н.Р.Николаев («Торунь»)* з.Фильченков («Зауралье») н.Д-а Андреев з.Вяч.Григорян (оба в «Академию Михайлова») з.Шаманин («Юниор») в.О.Д.Назаров («Дизель») н.А.Юшков («Горняк» Уч, через возврат аренды «Ак Барсу») з.Лесников («Юность-Минск») н.Арт.Гиндуллин з.Д.Вяткин з.О.Мисюль н.Дергунов н.Софьин («Алтай» У-К) з.Дроков
«Зауралье» Курган (фарм-соглашение с «Металлургом» Мг)
тренерский штаб: гм.Смагин гт.Альб.Логинов т.Андросов т.Н.Золотухин («Металлург» Жл) т.Вяч.Седов
——————
покинули тренерский штаб: т.В.Хромых («Сокол») т.Душкин («Барс»)
игроки остались: в.Ярославлев з.Сулимов з.Янчевский з.Ольшанский н.Джиг н.Т.Меньщиков н.Крутиков н.Сероух н.Авраменко н.Е.Коробкин н.Гарифулин н.Ал-р И Черников н.Шиафотдинов
——————
новые игроки: н.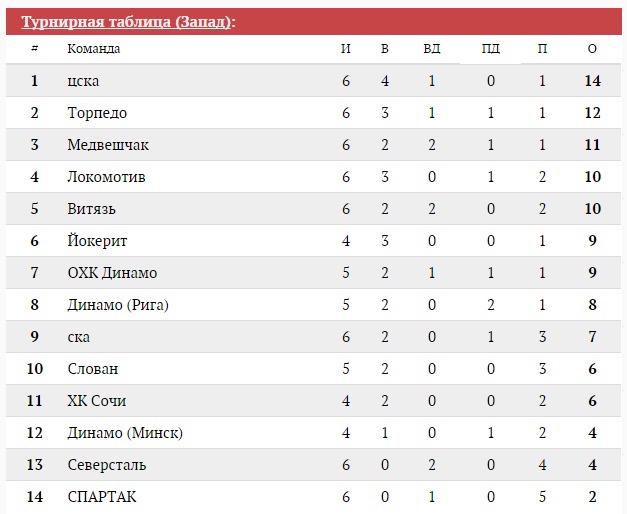 Дубин з.Г.Бабинцев н.Пятин (все из «Стальных Лисов») н.Рехтин («Рубин») з.Икамацких н.С.Жилов (оба из «Звезды») з.Дыньков («Тамбов») з.Е.Артамонов з.Семичастнов (оба из «Дизеля») з.Кудряшов («Иртыш») з.Фильченков («Ермак») н.Вад.Шутов («Химик») н.Коростин («Металлург» Нк) в.Устинский (экс-«Автомобилист») н.Глущенко («Крылья Советов») з.Минулин («Эверетт» WHL, через ММг) з.Дм.Ю.Зайцев («Металлург» Мг) в.Носов («Дизель», через «Металлург» Мг)
Дубин з.Г.Бабинцев н.Пятин (все из «Стальных Лисов») н.Рехтин («Рубин») з.Икамацких н.С.Жилов (оба из «Звезды») з.Дыньков («Тамбов») з.Е.Артамонов з.Семичастнов (оба из «Дизеля») з.Кудряшов («Иртыш») з.Фильченков («Ермак») н.Вад.Шутов («Химик») н.Коростин («Металлург» Нк) в.Устинский (экс-«Автомобилист») н.Глущенко («Крылья Советов») з.Минулин («Эверетт» WHL, через ММг) з.Дм.Ю.Зайцев («Металлург» Мг) в.Носов («Дизель», через «Металлург» Мг)
——————
игроки выбыли: н.Дюкарев («Тамбов») з.Коренков («Ермак») з.Игошев («Сокол» через ММг и «Амур») в.Старостин («Горняк» Уч) з.Г.Глебов («Химик», через «Спартак») з.Маккаев («Молот») з.Ю.Маслов («Буран») н.Ив.Исаев («Торпедо» У-К) н.И.Лисин («Адмирал», через возврат аренды «Локомотиву») н.Курепанов («Южный Урал») н.Сандер («Рубин») н.Колготин н.Ден.Цыганов (оба в «Юность-Минск») в.Нечистовский з.Гуляев (оба в «Хумо») н.Куликовский («Юниор») з.П.Володин («Дизель») н.А.Складниченко з.С.Гаврилов
«Звезда» Москва (фарм-клуб ЦСКА)
тренерский штаб: гт. Чебатуркин cт.Ег.Михайлов т.Жигарев т.В.Брызгалов мдж.Ден.В.Денисов (дебют)
Чебатуркин cт.Ег.Михайлов т.Жигарев т.В.Брызгалов мдж.Ден.В.Денисов (дебют)
——————
покинули тренерский штаб: мдж.А.Харламов («Торпедо» НН)
игроки остались: в.А.Самойлов в.Лозебников з.Луговяк з.Евг.Воронков з.Мясищев з.Биткин з.М-ль Ибрагимов н.А-м Антипов н.Силаев н.Усков н.Н.Е.Мокин н.Ег.Филин н.Н.О.Попугаев н.Ртищев н.Дан.Лобанов н.Каницкий н.М.Рыжков н.Комиссаров
——————
новые игроки: н.Замалиев н.Соркин (оба из «Красной Армии») н.С.Лапин («Лада») з.Д.Д.Юшкевич («Югра») н.Пешехонов («Сибирь», через ЦСКА) з.Калиниченко («Три-Сити» WHL, через ЦСКА) з.Е.Брютов («Рубин», через ЦСКА) н.Афонасьевский («Челмет», через ЦСКА) н.Пеньковский («Трактор») з.Чернюк («Торпедо» У-К, через «Алтай-Торпедо»)
——————
игроки выбыли: з.Чмыхов н.Толчинский (оба в ЦСКА) з.Анд.Ермаков («Сибирь», в аренду) з.Икамацких н.С.Жилов (оба в «Зауралье») з.А.Краснов («Ермак») з.Ф.Беляков («Горняк» Уч, через «Автомобилист») н.Карабань («КРС-БСУ») н.А.Хацей («Ижсталь») в. М.Третьяк («Динамо» Р) в.А.Корепанов («Лада») н.П.Зыков («Рубин») н.М.Мещеряков («Тамбов», в аренду) н.И.Левицкий («Торпедо-Горький», через «Торпедо» НН) н.Корбит н.Л.Климов (оба в «Красную Армию») з.Н.Макеев («Северсталь», через ЦСКА) з.Е.Цветков («Ростов») н.М.Бякин («Горняк» Уч) н.Н-а Назаров («Алтай-Торпедо») н.Подлубошнов
М.Третьяк («Динамо» Р) в.А.Корепанов («Лада») н.П.Зыков («Рубин») н.М.Мещеряков («Тамбов», в аренду) н.И.Левицкий («Торпедо-Горький», через «Торпедо» НН) н.Корбит н.Л.Климов (оба в «Красную Армию») з.Н.Макеев («Северсталь», через ЦСКА) з.Е.Цветков («Ростов») н.М.Бякин («Горняк» Уч) н.Н-а Назаров («Алтай-Торпедо») н.Подлубошнов
«Ижсталь» Ижевск (фарм-соглашение с «Авангардом»)
тренерский штаб: гт.Сайфуллин («Красная Армия») т.А.Первушин («Омские Ястребы») т.И.Кропотин (экс-гт) т.Митрошин
——————
покинули тренерский штаб: т.А.Вахрушев
игроки остались: в.Кононов в.Ф.Долганов з.Ал-й Виноградов з.Сухих з.Халиков з.Д-л Арефьев н.А.В.Баженов н.Рыжов н.Вал.Макаров н.Кочуров н.Кир.Афанасьев н.М.Кропотин з.Ал-р Козырев
——————
новые игроки: н.Ант.Ковалёв н.Н.Самохвалов н.Рожковский (все из «Югры», через АО) з.Щёкин з.Спиценко н.Шама н.М.Мельников н.Дмитричев (все из «Омских Ястребов») з.Шепельков з.Маркус (оба из «Красной Армии») н. Грибов («Саров») н.А.Хацей («Звезда») н.Евг.Ал.Орлов («Югра») н.Щегольков з.Дервук (оба из «Динамо» Пардубице, через АО) в.Костин («Торпедо» НН, через АО) н.Лаута (экс-«Сарыарка») н.М.Заседа («Авангард») н.Т.Манукян («Д.Дружковка») н.Мизюрин («Ньюфаундленд» ECHL через АО)
Грибов («Саров») н.А.Хацей («Звезда») н.Евг.Ал.Орлов («Югра») н.Щегольков з.Дервук (оба из «Динамо» Пардубице, через АО) в.Костин («Торпедо» НН, через АО) н.Лаута (экс-«Сарыарка») н.М.Заседа («Авангард») н.Т.Манукян («Д.Дружковка») н.Мизюрин («Ньюфаундленд» ECHL через АО)
——————
игроки выбыли: н.Н-а Вл Колесников н.Заплатников (оба в «Молот», через «Северсталь») н.Щанкин («Горняк» Рд) н.Ведерников («Торпедо-Горький») з.Ев.Борисов з.Топорков н.В.Горшков (все в «Ростов») н.В.Хабаров («Дизель») з.Курдюков («Ценг Тоу») з.Щенков («Динамо» Тв) з.Мерзляков («Юниор») з.Щёголев н.Семакин в.Гичев н.Д.Семенихин н.Ден.Иванов н.К.Черных з.Банцерев н.А-м Чупин
«КРС-БСУ» Пекин, China (фарм-клуб «Куньлуня»)
тренерский штаб: гт.Кампел (мдж.сб.США-U18) т.Игнатьев («Курбадс») гм.Оторбаев (администрация «Куньлуня»)
игроки: в.П.О’Брайен з.К.Джо з.П.Чжан н.Цзэц.Чжан н.Я.Ху н.Анд.Максимов (все из «КРС-ORG») н.Риггз н. Н-н Ли (оба из МХК «КРС Хэйлунцзян») з.Т.Шульц («Медисин Хит», WHL через КРС) н.Маури («Скалица», Словакия-2 через КРС) з.Чо («Джорджтаун», OJHL через КРС) н.Мэги (унив.Альберты, USports через КРС) з.М.Чен («Южная Каролина», ECHL через КРС) з.Чэнь (унив.Алабамы, NCAA через КРС) в.Ц.Сунь н.Сето н.Ин (все из «Куньлуня») н.Б.Вон (экс-«Хэйлунцзян», ВХЛ) н.Уолчак («Адирондак», ECHL через КРС) з.Салия («Попрад», через КРС) н.Ж.То-Ландри (возобновил карьеру) з.Ш.Ван («Ценг Тоу») з.Заборников («Торпедо» У-К, через КРС) н.Г.Скворцов («Динамо» Р, через КРС) н.Потехин («Витязь, через КРС) в.Мьюз («Уилинг», ECHL через КРС) з.Базевичс («Мейкон», SPHL через КРС) н.Карабань («Звезда», через КРС) н.М.Плотников (экс-«Юность-Минск»)
Н-н Ли (оба из МХК «КРС Хэйлунцзян») з.Т.Шульц («Медисин Хит», WHL через КРС) н.Маури («Скалица», Словакия-2 через КРС) з.Чо («Джорджтаун», OJHL через КРС) н.Мэги (унив.Альберты, USports через КРС) з.М.Чен («Южная Каролина», ECHL через КРС) з.Чэнь (унив.Алабамы, NCAA через КРС) в.Ц.Сунь н.Сето н.Ин (все из «Куньлуня») н.Б.Вон (экс-«Хэйлунцзян», ВХЛ) н.Уолчак («Адирондак», ECHL через КРС) з.Салия («Попрад», через КРС) н.Ж.То-Ландри (возобновил карьеру) з.Ш.Ван («Ценг Тоу») з.Заборников («Торпедо» У-К, через КРС) н.Г.Скворцов («Динамо» Р, через КРС) н.Потехин («Витязь, через КРС) в.Мьюз («Уилинг», ECHL через КРС) з.Базевичс («Мейкон», SPHL через КРС) н.Карабань («Звезда», через КРС) н.М.Плотников (экс-«Юность-Минск»)
«Лада» Тольятти
тренерский штаб: сп.дир.Кречин гт.Ал-р Н Титов т.Анат.Степанов (оба из «Сокола») т.О.Н.Волков (ДЮСШ «Лада») т.Вл.С.Волков (экс-«Торос») т.Н.Титов (дебют)
——————
покинули тренерский штаб: гт.Анат. Емелин т.А.Г.Нестеров («Молот») т.Арк.Андреев т.Курошин т.С.Медведев («Торос»)
Емелин т.А.Г.Нестеров («Молот») т.Арк.Андреев т.Курошин т.С.Медведев («Торос»)
игроки остались: в.Скрынник з.Усманов з.Возовик з.Бердюков з.Д.Семёнов н.Кизимов н.Андрющенко н.А.Озолиньш н.К.Майоров н.Тимирёв н.Мастрюков н.Кутявин н.А-м Артёмов
——————
новые игроки: н.Н.Лукин («Ценг Тоу») н.В.Журавлёв з.Рин.Шафигуллин з.Т.Булатов (все из ЦСК ВВС) з.Чурляев з.Капотов н.Ю.Назаров (все из «Сокола») з.Кротиков («Рубин») н.А.Денежкин («Динамо» СПб) в.А.Корепанов («Звезда») з.Валиуллов («Ладья») н.Толузаков («КРС-ORG») н.Шураков («Сокол», через «Торпедо» НН) н.И.Угольников (ЦСК ВВС, через «Нефтехимик») з.Крылов («Сокол», через «Спартак»)
——————
игроки выбыли: н.С.Лапин («Звезда») з.Постников н.Н.Владимиров (оба в «Сокол») н.А.Гришаков («Трактор») з.Костромитин («Хёрнинг») з.Ф.Метлюк (завершил игровую карьеру) з.Дюков («Динамо» Мн) н.И.Романов («Буран», через возврат аренды «Локомотиву») н.Бондырев («Нефтяник») н.Ег.Ал.Алексеев («Чайка») в.Арс.Никоноров («Кристалл» С) з. Р.Дубинин («Тамбов») н.Сигарёв («Югра») н.Куваев («Молот») н.И.Безруков («Хумо») н.Головкин з.Ев.Курбатов н.С.Чистов з.Ал.Платонов в.Магарилов («Гомель»)
Р.Дубинин («Тамбов») н.Сигарёв («Югра») н.Куваев («Молот») н.И.Безруков («Хумо») н.Головкин з.Ев.Курбатов н.С.Чистов з.Ал.Платонов в.Магарилов («Гомель»)
«Металлург» Новокузнецк (фарм-соглашение с «Сибирью»)
тренерский штаб: гт.Д.Пархоменко т.С.Бердников т.Мишин ио.сп.дир.Турукин (дебют)
——————
покинули тренерский штаб: т.Сыченко («Динамо» Тв)
игроки остались: з.Щукин з.Ег.Жданов н.Дюрягин н.Шубин н.Ал-р А Шевченко н.Ердаков н.В.Поляков н.Ал-р Локтев
——————
новые игроки: в.А.Литвинов н.Малетин (оба из «Сокола», возврат аренды) з.Акишин («Тамбов») н.А-м С Чернов («Торос») з.Aл-й Е Kириллов («Химик») в.Печурский («Торпедо» У-К) з.Э.Шевченко з.Д.Галеев (оба из «Югры») з.Юксеев («СКА-Нева») н.Сарваров н.М.Кицын н.С.Бутузов (все из «Сарыарки») з.Ив.Глазков («Северсталь») н.Лудцев (ЦСК ВВС) н.К.Пархоменко («Юниор») н.А.Рубан («Алтай-Торпедо») н.С.Юдин («Кузнецкие Медведи») н.Зырянов («Сибирь») з.Рябоконов («Южный Урал»)
——————
игроки выбыли: з. Воробей («Пеликанс») н.Сем.Иванов з.С.Калашников (оба в «Рубин») н.А.Гловацкий («Сокол») в.В.Гросс («Автомобилист», возврат аренды) н.Ег.Морозов в.Р.Хакимов з.Балдаев (все в «Северсталь») з.К.Маслов («Молот», через «Северсталь») н.Коростин («Зауралье») в.Гафиуллин («Дизель», в аренду) н.Скобелев («Сарыарка») н.Д.Жукенов («Горняк» Уч) з.Е.Ерёменко з.Швалёв («Металлург» Жл) н.Пав.В.Медведев («Дизель») н.С.Барбашев («СКА-Нева», через СКА) н.Земченко («Молот») н.Сатаров («Кузнецкие Медведи») з.Виксна з.Скутар з.Ал-р С Макаров
Воробей («Пеликанс») н.Сем.Иванов з.С.Калашников (оба в «Рубин») н.А.Гловацкий («Сокол») в.В.Гросс («Автомобилист», возврат аренды) н.Ег.Морозов в.Р.Хакимов з.Балдаев (все в «Северсталь») з.К.Маслов («Молот», через «Северсталь») н.Коростин («Зауралье») в.Гафиуллин («Дизель», в аренду) н.Скобелев («Сарыарка») н.Д.Жукенов («Горняк» Уч) з.Е.Ерёменко з.Швалёв («Металлург» Жл) н.Пав.В.Медведев («Дизель») н.С.Барбашев («СКА-Нева», через СКА) н.Земченко («Молот») н.Сатаров («Кузнецкие Медведи») з.Виксна з.Скутар з.Ал-р С Макаров
«Молот-Прикамье» Пермь (фарм-соглашение с «Северсталью»)
тренерский штаб: гт.Десятков т.А.Ершов т.Меркесов (все из «Рязани») т.А.Г.Нестеров («Молот»)
——————
покинули тренерский штаб: гт.В.Долишня т.Д.Кокорев (оба в «Капитан») т.Д.Черепанов
игроки остались:
——————
новые игроки: в.Шугаев н.Жирнов н.А-м Марков (все из «Алмаза») з.С.Тунхузин н.Вл.Королёв (оба из «Рубина») з.Е.Калинин («Ценг Тоу») в. М.Шпаков з.Щербов з.Вал.Ю.Васильев з.Седунов з.Д.Мнацян н.Чувиляев н.Дм.Ю.Воробьёв н.А-м Прохоров н.Луговой (все из «Рязани») з.Н.Зеленин («Дизель») н.Ден.Фахрутдинов н.Сметанин н.Д.Скатов (все из «Южного Урала») н.Заплатников н.Н-а Вл Колесников (оба из «Ижстали», через «Северсталь») н.В.Глебов («Крылья Советов») н.Зенчиков («Локо») з.Маккаев («Зауралье») з.В.Сычушкин н.И.Захарчук н.С.Купцов (все из «Северстали») з.К.Маслов («Металлург» Нк, через «Северсталь») н.Анд.Ал.Белозёров («Торпедо» У-К, через «Северсталь») н.Куваев («Лада») н.Земченко («Металлург» Нк)
М.Шпаков з.Щербов з.Вал.Ю.Васильев з.Седунов з.Д.Мнацян н.Чувиляев н.Дм.Ю.Воробьёв н.А-м Прохоров н.Луговой (все из «Рязани») з.Н.Зеленин («Дизель») н.Ден.Фахрутдинов н.Сметанин н.Д.Скатов (все из «Южного Урала») н.Заплатников н.Н-а Вл Колесников (оба из «Ижстали», через «Северсталь») н.В.Глебов («Крылья Советов») н.Зенчиков («Локо») з.Маккаев («Зауралье») з.В.Сычушкин н.И.Захарчук н.С.Купцов (все из «Северстали») з.К.Маслов («Металлург» Нк, через «Северсталь») н.Анд.Ал.Белозёров («Торпедо» У-К, через «Северсталь») н.Куваев («Лада») н.Земченко («Металлург» Нк)
——————
игроки выбыли: з.А.Ерохин («Хумо») з.С.Дубовицкий («Сахалинские Акулы») з.Ег.Сорокин (ЦСК ВВС) н.М-м Волков («Иртыш») н.Ил.А.Серегин («Гомель») н.Дельнов («Ермак», через «Адмирал») н.Градусов з.Ельшанский з.Шикун н.Тымченко н.Г.Сергеенко (все в «Ермак») н.А.Гомоляко («Торпедо-Горький», через «Сочи» и «Торпедо» НН) з.Н.Синицын («Арлан») н.Мих.О.Попов («Кулагер») н.Андриянов н.С.В.Абрамов (оба в «Дизель») н. Шайхулов («Нефтяник») з.Ив.Кузнецов («Тамбов») н.Тумченко («Ирбис», через «Ак Барс») з.В.Лысенко («Ценг Тоу») н.Слепынин («АК59») з.Ал-р Вал Никитин («Юниор») н.Ал-р Лебедев («Металлург» Жл) в.Анаркулов («Буран») в.Енюшин н.Анат.Васильев в.Летуновский н.Мокрицын з.Халемин
Шайхулов («Нефтяник») з.Ив.Кузнецов («Тамбов») н.Тумченко («Ирбис», через «Ак Барс») з.В.Лысенко («Ценг Тоу») н.Слепынин («АК59») з.Ал-р Вал Никитин («Юниор») н.Ал-р Лебедев («Металлург» Жл) в.Анаркулов («Буран») в.Енюшин н.Анат.Васильев в.Летуновский н.Мокрицын з.Халемин
«Нефтяник» Альметьевск (общая фин.структура с «Ак Барсом»)
тренерский штаб: гт.И.Гизатуллин т.А.Шахворостов т.Е.Мухин т.Ячанов т.Ураков
игроки остались: з.Салахов (аренда от «Ак Барса») з.Д.Хамидуллин з.Мулюков з.Сумин з.А.Сорокин з.Теряев н.Ястребков н.М.Мокин н.Ден.Ляпустин н.Л.Мансуров н.Д.Насыбуллин н.Вит.Каменев н.К.Лебедев н.С.Альшевский н.Я.Альшевский
——————
новые игроки: н.Ив.С.Иванов («Рязань») н.Шайхулов («Молот») в.Андрюхов («Франкфурт») з.Аблаев («Горняк» Уч) в.С.Коробов («Северсталь») н.Бондырев («Лада») з.Т.Фаткуллин («Барс», аренда) н.Мих.Шестопалов («Ирбис», аренда) в.Минегалиев з.Аввакумов н.Т.Закиров (все из «Спутника» А) з.Патрикеев («Адмирал») н. Е.Баранов («Слован»)
Е.Баранов («Слован»)
——————
игроки выбыли: н.Д-а Цыганов («Буран») н.Анд.В.Демидов («Иртыш») в.Филоненко («Южный Урал», через «Звезду») в.И.Голубев з.О.Железнов н.Хапов з.Адамчук н.Д.Воеводин (все в «Барс», через «АБ») з.Э.Янсонс («Динамо» Р) н.Ядроец («Рубин») в.Смирягин («Хумо») н.У.Гильманов з.В.Арбузов (оба в «Ценг Тоу») н.А-м Осипов («Рубин») н.Н.Елисеев
«Номад» Нур-Султан, Казахстан (фарм-клуб «Барыса»)
тренерский штаб: гт.Ю.Михайлис ст.Г.Верещагин т.Бессонов сп.дир.Мамбеталиев (Казахстан-U16)
игроки остались: в.Крамарь в.Стрия з.Бакушин з.Рыспаев з.Пелевин з.Маржикпаев н.Анд.Яковлев н.Преснов н.Ив.Г.Верещагин н.Д.Макеев н.Рунов н.Шмурыгин н.Хасынханов н.Гурин
———————
новые игроки: в.С.Кудрявцев з.Ибрайбеков н.Асетов н.Я.Евдокимов н.Гурков (все из «Барыса») з.С-т Данияр н.Гатиятов н.С-н Данияр н.М.Мухаметов н.Б.Муратов (все из «Снежных Барсов») з.Ст.Александров («Алтай-Торпедо») н.Муравьёв («Ирбис») н. Некряч н.Ал.Скабелка (оба из «Бейбарыса») з.Бухряков («СКА-1946») н.Жайлауов («Алтай-Торпедо», через «Барыс») н.Разумняк (ЦСК ВВС) з.Костарев («Челмет») з.Дубовик («Горняк» Уч)
Некряч н.Ал.Скабелка (оба из «Бейбарыса») з.Бухряков («СКА-1946») н.Жайлауов («Алтай-Торпедо», через «Барыс») н.Разумняк (ЦСК ВВС) з.Костарев («Челмет») з.Дубовик («Горняк» Уч)
———————
игроки выбыли: в.Мальгин з.Лакиза з.Яхонт з.Долгушев н.Ковзалов н.Вяч.Ипатов (все в «Алматы») н.Королинский («Темиртау») з.Клещенко («Барыс») н.Савицкий («Торпедо» У-К) н.Ал-р Мелихов («Алтай-Торпедо») з.Церенок («Актобе») н.Д.Габдуллин («Бейбарыс») н.Сейтанов («Ценг Тоу»)
«ORG» Пекин, China (фарм-соглашение с «Куньлунем»)
тренерский штаб: гт.Грэхэм («Форт-Уэйн», ECHL) т.Дм.Катаев («Беларусь U18») т.Г.Ван (МХК «КРС Хэйлунзян») т.Гратц (экс-«Гринвилл», ECHL)
——————
покинули тренерский штаб: гт.А.Э.Барков т.Сарматин т.И.О.Горбенко_93гр т.Пуурула (все в «ORG Юниор»)
игроки остались: в.Лием в.Дан в.Боярчук з.Рыбницкий з.Полинин з.Т.Ху з.Цзяц.Чжан н.П.Хуан н.Ж.Цзо н.Х.Чжан н.К.Макаров н.Т.Цзо н.Ц.Хуан н. Мих.А.Абрамов
Мих.А.Абрамов
——————
новые игроки: н.П.Цзи (экс-«Ценг Тоу») н.Т.Ся (сборная China) н.Чжу (экс-«Харбин») н.Ц.Ван (МХК «КРС Хэйлунцзян») н.Тянулин («Брамптон», ECHL) з.И.Неколенко («Ньюфаундленд», ECHL) з.Морулёв («Алмаз») н.Михасёнок («Бейбарыс») н.С.Столяров («Алтай» У-К) з.М.Мишаков («Стальные Лисы») н.Г.Дежарден («Сендерюск», через КРС) н.Джаспер («Фредериксхавн», через КРС) н/з.Р.Шин (унив.Калгари, USports через КРС) з.Ши (экс-«Форт-Уэйн», ECHL через КРС) з.Э.Насыбуллин («Барс») н.Забабурин («Саров») з.Швиденко («Сокол») н.С.Бабинцев («Ньюфаундленд», ECHL)
——————
игроки выбыли: н.Толузаков («Лада») з.Дэх.Чжан н.Х.У (оба в «Ценг Тоу») в.П.О’Брайен з.К.Джо з.П.Чжан н.Т.Цзо н.Ц.Хуан н.Анд.Максимов н.Хаген н.Цзэц.Чжан н.Я.Ху (все в «КРС-БСУ») з.Там («Куньлунь») н.Крутий («Сарыарка») н.Ил.А.Орлов з.Ю.Хань н.Сян з.Цуриков («Темиртау»)
«Ростов» Ростов-на-Дону
тренерский штаб: гт.Г.Пантелеев тр/спорт.дир.Т. Кулль т.С.Сухарев
Кулль т.С.Сухарев
игроки остались: з.Шевчук з.Ал-й В Царёв з.Шулев з.Мих.А.Степанов н.В.Туник н.Мякинин н.Анд.И.Мартынов н.А.Коробов н.Щербаков н.Н.Рогов н.Е.Кулаков н.Анд.Леонов н.Опалев
——————
новые игроки: н.Г.Егоров в.Кудашев (оба из «Юниора») в.Н.Масленников («Сарматы») н.Анд.О.Алексеев («Саров») М.Субботин («Мордовия») в.С.А.Павлов («Кристалл» С) Евг.Борисов з.Топорков н.В.Горшков (все из «Ижстали») н.Ег.Кузьменко («Химик») н.Миргалиев («Темиртау») з.Басистый («СКА-1946») з.Е.Цветков («Звезда») з.А.Антонов («Дизель»)
——————
игроки выбыли: н.Ал-й Прохоров («Южный Урал») н.Пукс («Кременчуг») н.Гиберт з.М.Блинов (оба в «Динамо-Алтай») з.Шуркалов в.Кукурудза н.Пав.А.Медведев з.Ив.Комаров з.Э.Хасанов з.И.Меркулов н.Поляничев з.Н.Глотов
«Рубин» Тюмень
тренерский штаб: гт.М.Звягин т.А.Исаков т.Кир.А.Брагин т.Долгушин (экс-«Тайфун») гм.Н.Бабенко
——————
покинули тренерский штаб: т. К.Власов («Автомобилист»)
К.Власов («Автомобилист»)
игроки остались: в.Е.Назаров з.Ал.А.Федотов з.Кунгурцев з.К.Фаст з.Ал-р М Родионов н.Ден.Ал.Давыдов н.К.Белов н.В.Кравченко н.Д.Ячменёв н.Ант.Угольников н.И.Степанов
——————
новые игроки: з.Ал-р Е Ильин н.Сусликов н.А.Тимофеев (все из «Тюменского Легиона») н.Сандер («Зауралье») н.Мнихович (ЦСК ВВС) н.Сем.Иванов з.С.Калашников (оба из «Металлурга» Нк) з.Дм.Лютов н.М.Железнов (оба из «Сарыарки») н.П.Зыков («Звезда») н.Чугуев («Южный Урал») в.Артамкин («Северсталь») з.Анд.Н.Алексеев («Ценг Тоу») н.Ядроец («Нефтяник») з.В.Зотов («Сахалин») з.Петриков («Челмет», через аренду из «Трактора») н.Р.Горбунов («Шахтёр» С) н.Н.Складниченко («Торпедо» У-К)
——————
игроки выбыли: н.Байкеев («Дизель») н.В.Коротков («Сокол») з.Верёвкин («Югра») н.Д.Бойчук н.Комаристый (оба в «Сарыарку») н.Ил.Карлин («Химик») з.С.Козлов («Горняк» Уч) з.Ал-й Кожевников («Южный Урал») з.Кротиков («Лада») н.Лешков («Чебоксары») н.Рехтин («Зауралье») з.С. Тунхузин н.Вл.Королёв (оба в «Молот») н.Чиглинцев н.А.Князев-ст (оба в «Буран») з.Е.Брютов («Звезда», через ЦСКА) в.Тяло н.Ег.А.Попов (оба в «Динамо» Тв, через ДМо) з.М.Богданов н.Н.Брютов в.П.Гущин (все в «Тюменский Легион») з.Лисов («Торпедо-Горький», через «Торпедо» НН) н.Мих.Шабанов («Оренбург»)
Тунхузин н.Вл.Королёв (оба в «Молот») н.Чиглинцев н.А.Князев-ст (оба в «Буран») з.Е.Брютов («Звезда», через ЦСКА) в.Тяло н.Ег.А.Попов (оба в «Динамо» Тв, через ДМо) з.М.Богданов н.Н.Брютов в.П.Гущин (все в «Тюменский Легион») з.Лисов («Торпедо-Горький», через «Торпедо» НН) н.Мих.Шабанов («Оренбург»)
ХК «Рязань» Рязань
тренерский штаб: гт.А.Г.Сырцов («Химик») т.Д.Евстигнеев (экс-«Атланты»)
——————
покинул тренерский штаб: гт.П.Н.Десятков т.А.Ершов т.Меркесов (все в «Молот») гм.В.И.Нарзяев («Тамбов»)
игроки остались: н.Вл-р Карпов в.С.Денисов н.С.Мельников
——————
новые игроки: н.Муштаев н.Ал-р Воронин (оба из «Алматы») з.В.Демаков («Горняк» Рд) з.Р.Осипов («Ермак») н.С.Третьяков («Горняк» У) з.Анд.С.Миронов н.И.Каштанов (оба из «Сарова») з.Е.Шакуров («Торос») в.С.Большаков («Химик») н.Чванчиков («Тамбов») н.Кудрин («Южный Урал») з.А.Яценко («Пинские Ястребы») н.П.Злобин («Полёт») н.Вл-в Волков («Дизель») з. К.Лазарев («Спутник» Ал) н.Закурин («Мордовия») н.А.Кокшаров («Крылья Советов») н.З.Чернов («Алтай» Б) з.Е.Дубровин («Иртыш») н.Жердев (экс-«Динамо» Р) з.Горечишников з.Моренец (оба из «Горняка» Рд) н.Ал-й Филиппов (экс-«Молот») н.А-м Шакиров («Атланты») з.Н-а Поляков («Дизелист») з.Харькин (экс-«Ермак») н.А-м Мелихов («Челмет»)
К.Лазарев («Спутник» Ал) н.Закурин («Мордовия») н.А.Кокшаров («Крылья Советов») н.З.Чернов («Алтай» Б) з.Е.Дубровин («Иртыш») н.Жердев (экс-«Динамо» Р) з.Горечишников з.Моренец (оба из «Горняка» Рд) н.Ал-й Филиппов (экс-«Молот») н.А-м Шакиров («Атланты») з.Н-а Поляков («Дизелист») з.Харькин (экс-«Ермак») н.А-м Мелихов («Челмет»)
——————
игроки выбыли: в.П.Кочетков (СКА, через «Сочи») з.Колегов з.О.Попов н.Э.Васильев з.А.Большаков (все в «Тамбов») н.К.Беляев («Торпедо-Горький») н.В.В.Нарзяев («Тамбов», через «Сочи») в.Пиманкин («Горняк» Рд) в.М.Шпаков з.Вал.Ю.Васильев з.Щербов з.Д.Мнацян з.Седунов н.Чувиляев н.Дм.Ю.Воробьёв н.А-м Прохоров н.Луговой (все в «Молот») н.Ал.Репьях з.Н-а Е Попов (оба в «Сокол») н.Рукин з.П.П.Десятков (оба в ЦСК ВВС) н.Шацкий («Буран») н.Ив.С.Иванов («Нефтяник») н.Ив.Анд.Иванов («Динамо» Тв) н.Ворошило («Хумо») н.Ал-р И Васильев («Д.Дружковка») в.Кожокарь
«Сарыарка» Караганда, Казахстан
тренерский штаб: гт. Л.Тамбиев т.Банада т.Акифьев т.С.С.Белов (МХК «Динамо СПб») гм.Ал-й И Кузнецов (экс-нач.ком)
Л.Тамбиев т.Банада т.Акифьев т.С.С.Белов (МХК «Динамо СПб») гм.Ал-й И Кузнецов (экс-нач.ком)
——————
покинули тренерский штаб: т.Костючёнок («Югра») т.Трибунцов («Динабург»)
игроки остались: в.Рейзвих в.Бояркин з.Ант.Горбачёв з.Лыпкань з.Чёботов з.Кропачёв з.Сиксна н/з.Э.Нургалиев н.Валицкий н.Ремов н.А.Борисевич н.Зеленков н.Вл.Никулин
——————
новые игроки: в.Д.Еремеев («Снежные Барсы») з.С.А.Кузнецов н.Дм.Э.Михайлов н.Голоднюк (все из «Югры») з.Ив.А.Воробьёв з.П.Лукин н.К.Кузнецов н.Анд.Бирюков (все из «Динамо» СПб) н.Д.Бойчук н.Комаристый (оба из «Рубина») н.Галоха («Нейи-сюр-Марн», Франция D2) н.Скобелев («Металлург» Нк) н.Закарлюкин («Сокол») н.Чапоров («Юность U18», Караганда) н.Крутий («КРС-ORG») н.О.Яшин («Катовице») н.Д.Марушев («Ирбис») н.Кокуёв («Трактор») н.Панков («Торос») н.В.Москалёв («Дизель»)
——————
игроки выбыли: з.Ег.Наместников («Иртыш») з.Метальников («Барыс») з.Дм.Лютов н.М.Железнов (оба в «Рубин») н. Сарваров н.М.Кицын н.С.Бутузов (все в «Металлург» Нк) н.Зиазов н.Махановский н.Ряшенцев (все в «Югру») н.К.Соколов («Сочи») з.Ибатуллин н.Ломако (оба в «Адмирал») н.Желнеровский («Юность-Минск») н.А.Буяльский з.В.Шульга (оба в «Темиртау») н.К.Тамбиев («Курбадс»)
Сарваров н.М.Кицын н.С.Бутузов (все в «Металлург» Нк) н.Зиазов н.Махановский н.Ряшенцев (все в «Югру») н.К.Соколов («Сочи») з.Ибатуллин н.Ломако (оба в «Адмирал») н.Желнеровский («Юность-Минск») н.А.Буяльский з.В.Шульга (оба в «Темиртау») н.К.Тамбиев («Курбадс»)
«СКА-Нева» Санкт-Петербург (фарм-клуб СКА)
тренерский штаб: гт.К.Курашёв («Кур») т.М.В.Милехин («СКА-Варяги») т.А.Парфёнов (МХК КРС «Хэйлунцзян») т.П.Орлов т.Якимович
——————
покинули тренерский штаб: гт.Кравец («Витязь») т.И.Ефимов (СКА) т.Ал-й П Воробьёв («СКА-Варяги»)
игроки остались: в.Лысенков з.Холоденин з.Солянников з.М.Тихонов з.Калабушкин з.Н.Глухов з.Валенцов з.В.Сёмин н.Конозов н.И.Володин н.Петьков н.Полодян н.В.Курбатов н.Ожгихин н.Цицюра н.Кукштель н.А-м Николаев н.Д.Овечкин н.Ю.Скворцов н.Огирчук н.Кир.Марченко
——————
новые игроки: в.М.Гибадуллин з.Д-а Арефьев з.Д.Баринов н.Царюк (все из «СКА-1946») в.Я.Аскаров н.Яровинский (оба из «СКА-Варягов») н. Савунов («Дизель», через СКА) н.Ив.Морозов (СКА) з.Мироманов («Манчестер» ECHL через СКА) н.С.Барбашев («Металлург» Нк, через СКА) н.В.Глотов («Цинциннати» ECHL через СКА)
Савунов («Дизель», через СКА) н.Ив.Морозов (СКА) з.Мироманов («Манчестер» ECHL через СКА) н.С.Барбашев («Металлург» Нк, через СКА) н.В.Глотов («Цинциннати» ECHL через СКА)
——————
игроки выбыли: в.Н.Богданов («Тамбов», через «Сочи») н.Орлович-Грудков («Сочи») н.Подколзин в.А.Мельничук з.Галенюк (все в СКА) н.А.Цыплаков н.Ник.Поляков з.Д.Семыкин н.Жук (все в «СКА-1946») н.Щербина н.Е.Григоренко (оба в «Хумо») в.Ал-й Макс Иванов («Динамо-Молодечно») з.Юксеев («Металлург» Нк) н.Св.Гребенщиков н.Ларичев н.Дан.Квартальнов (все в «Витязь») з.Кныжов («Сан-Хосе») н.Шарпанских
«Сокол» Красноярск (фарм-соглашение с «Амуром»)
тренерский штаб: гт.Хромых («Зауралье») ст/гм.Никишов т.Баев («Юниор») т.Кривомазов
——————
покинули тренерский штаб: гт.Ал-р Н Титов т.Анат.Степанов (оба в «Ладу»)
игроки остались: в.Е.Киселёв з.Колганов з.А.Савельев з.Е.Журавлёв н/з.Дм.Цыганов н.Вилков н.Мисников н.Г.Мищенко н.Ал-й Князев-мл н. Гиздатуллин н.Д.Горбунов н.Болтанов
Гиздатуллин н.Д.Горбунов н.Болтанов
——————
новые игроки: н.А.Гловацкий («Металлург» Нк) н.Ал.Репьях з.Н-а Е Попов (оба из «Рязани») в.Шилин («Югра») з.Постников н.Н.Владимиров (оба из «Лады») н.В.Коротков («Рубин») з.Бочков («Торос») з.Игошев («Зауралье», через ММг и «Амур») н.Чехов («Красноярские Рыси») з.Камалов («Амур») н.Ег.Лебедев («Ценг Тоу», через аренду от «Югры») з.Пав.С.Медведев («Торпедо-Горький», через ТНН и «Амур»)
——————
игроки выбыли: в.А.Литвинов н.Малетин (оба в «Металлург» Нк, возврат аренды) н.Симаков (тренер в «Горняке» Уч) з.Меляков («Торпедо» НН, возврат аренды) н.Шураков («Лада», через возврат аренды «Торпедо» НН) з.Чурляев з.Капотов н.Ю.Назаров (все в «Ладу») з.Крылов («Лада», через «Спартак») н.Закарлюкин («Сарыарка») з.В.Тесленко («Химик») н.В.Шевченко («Торос», через возврат аренды в «С.Юлаев») н.М-м Мальцев («Юность-Минск») з.Д.Григоркевич н.Бурмаго н.Потылицын (все в «Красноярские Рыси») в.Ю.В.Петров («Шахтёр» С) н.Берестенников («Торпедо-Горький», через «Амур» и «Торпедо» НН) з. Швиденко («ORG») з.С.Букарев («Земгале»)
Швиденко («ORG») з.С.Букарев («Земгале»)
ХК «Тамбов» Тамбов (фарм-соглашение с ХК «Сочи»)
тренерский штаб: гт.Прокопьев («Витязь») т.Решетников (экс-«U18», МХЛ) т.Анд.В.Шутов т.Стрыков гм.В.И.Нарзяев («Рязань»)
——————
покинули тренерский штаб: гт.Лунёв (МХК «Атлант») т.Вал.Олейник
игроки остались: в.Н.Ложкин в.Давлетшин з.Р.Кривошеев н.Шингареев н.М.Гимбатов н.А.Гимбатов з.А.Игнатов н.А.Клочков н.А.Воронков н.Вас.Жилов
——————
новые игроки: н.Ковальский (МХК «Липецк») н.Ревацкий («Горняк» У) з.О.Попов з.Колегов н.Э.Васильев з.А.Большаков (все из «Рязани») з.В.Репин («Южный Урал») н.М.Мещеряков («Звезда», в аренду) з.Ив.Кузнецов («Молот») з.Слепцов («Буран») н.В.В.Нарзяев («Рязань», через «Сочи») н.Селютин («Тиллсонбург», GMHL) н.Игнатушкин («Кошице») з.Р.Дубинин («Лада») н.Дюкарев («Зауралье») н.Басков («Дунареа») в.Н.Богданов («СКА-Нева», через «Сочи») з.А-м С Мальцев («Сочи»)
——————
игроки выбыли: з. Дыньков («Зауралье») з.Ал-р С Родионов («Адмирал») н.В.Коньков («Динамо-Алтай») н.Крашенинников («Торос») з.Акишин («Металлург» Нк) з.Вл-р Марченко («Дизель») н.Чванчиков («Рязань») з.Павликов («Торпедо-Горький», через «Локомотив» и «Торпедо» НН) н.Ал-й Ефимов («Алматы») н.Н.Свинцицкий (ЦСК ВВС) з.Яценков (завершил игровую карьеру) н.Дм.А.Орлов («Кременчуг») н.Ногин («Челны») в.Югов (МХК «Рязань») н.М.Жолобов н.Лощенко з.Томкин
Дыньков («Зауралье») з.Ал-р С Родионов («Адмирал») н.В.Коньков («Динамо-Алтай») н.Крашенинников («Торос») з.Акишин («Металлург» Нк) з.Вл-р Марченко («Дизель») н.Чванчиков («Рязань») з.Павликов («Торпедо-Горький», через «Локомотив» и «Торпедо» НН) н.Ал-й Ефимов («Алматы») н.Н.Свинцицкий (ЦСК ВВС) з.Яценков (завершил игровую карьеру) н.Дм.А.Орлов («Кременчуг») н.Ногин («Челны») в.Югов (МХК «Рязань») н.М.Жолобов н.Лощенко з.Томкин
«Торос» Нефтекамск (фарм-клуб «Салавата Юлаева»)
тренерский штаб: гт.Р.Сулейманов т.Ал.Гареев («Толпар») т.Мазитов т.С.Медведев («Лада»)
——————
покинули тренерский штаб: т.Ден.Хлыстов («Толпар») т.Рябков («Сибирь») гм.Дм.Денисов
игроки остались: з.К.Цулыгин з.Н.Воронин з.А.Банников з.Рубинчик з.Петрищев н.Пименов н.Бобылёв н.Чебыкин н.Пачин н.Е.Дубровский н.С.Емелин н.Факиров н.С.Голованов н.Г.Кузьмин н.И.Г.Баранов
——————
новые игроки: в.Брагинский в.Ю.Грошев з.Абдеев з.Анохин з. Варлов н.Пустозёров н.Д.Башкиров (все из «Толпара») з.Жульдиков («Челмет» через СЮ) н.Крашенинников («Тамбов») н.Анкудинов («Химик») н.Скориков н.В.Лукин (оба из «С.Юлаева») н.В.Шевченко («Сокол», через СЮ) з.Галин (IPK, Финляндия-2) в.Сохатский («Автомобилист», через СЮ)
Варлов н.Пустозёров н.Д.Башкиров (все из «Толпара») з.Жульдиков («Челмет» через СЮ) н.Крашенинников («Тамбов») н.Анкудинов («Химик») н.Скориков н.В.Лукин (оба из «С.Юлаева») н.В.Шевченко («Сокол», через СЮ) з.Галин (IPK, Финляндия-2) в.Сохатский («Автомобилист», через СЮ)
——————
игроки выбыли: н.А-м С Чернов («Металлург» Нк) н.Шэн («Бостон Брюинз») з.Бочков («Сокол») з.Грязев («Динамо» Тв) з.Е.Шакуров («Рязань») з.М.Рогов («Югра») в.Д-л Вад Тарасов («Эссет») в.И.Федотов («Трактор») н.Н.Ли («Дизель») н.А-м Гордеев в.Циркуль н.Н.Филатов н.Панков («Сарыарка»)
«Торпедо» Усть-Каменогорск
тренерский штаб: гт.А.Ждахин т.Бульин (оба из «Югры») Е.Фадеев (экс-гт) т.Е.Черепанов зам.гд.Быстрянцев (экс-мдж)
——————
покинули тренерский штаб: т.М.Ждахин (видеооператор)
игроки остались: в.М-м Сидоров з.Анд.Виноградов з.Ил.Антоновский з.Стулов н.Куракин н.П.Ждахин н.Лихотников н.Анд.Караваев н.Шин н.Кир.Полозов н.Рымарев
——————
новые игроки: н. Савицкий («Номад») н.С.Шестаков з.Молодцов (оба из «Динамо» СПб) н.Ал-р Тарасов («Дизель») н.Игнашин («Югра») н.Ив.Исаев («Зауралье») з.Д.Громов в.Перевозчиков з.В.Садиков (все из «Ценг Тоу») н.Гренц («Барыс») з.Полухин (МХК «КРС Хэйлунцзян») н.Н.Сироткин з.Таратунин (оба из «Адмирала») з.В.Ершов («Толпар») з.Д.Быков («Алтай-Торпедо»)
Савицкий («Номад») н.С.Шестаков з.Молодцов (оба из «Динамо» СПб) н.Ал-р Тарасов («Дизель») н.Игнашин («Югра») н.Ив.Исаев («Зауралье») з.Д.Громов в.Перевозчиков з.В.Садиков (все из «Ценг Тоу») н.Гренц («Барыс») з.Полухин (МХК «КРС Хэйлунцзян») н.Н.Сироткин з.Таратунин (оба из «Адмирала») з.В.Ершов («Толпар») з.Д.Быков («Алтай-Торпедо»)
——————
игроки выбыли: в.Печурский («Металлург» Нк) н.Анд.Ал.Белозёров («Молот», через «Северсталь») з.Заборников («КРС-БСУ», через «Куньлунь») з.Ивашин («Актобе») з.Якименко («Торпедо-Горький») з.С.Ильин («Хумо») н.Белухин («Д.Дружковка») н.М.Капитуров («Амур») н.Арк.Шестаков («Барыс») н.Д.Каштанов (ЦСК ВВС) з.Чернюк («Звезда», через «Алтай-Торпедо») з.Бекетаев н.Ал-р Меньщиков н.Епишев (все в «Алтай-Торпедо») з.А.Нурек («Лида») н.Шантыка з.Р.Хисамутдинов* н.Н.Складниченко («Рубин») в.А.Гаврилов
«Торпедо-Горький» Нижний Новгород (фарм-клуб «Торпедо» НН)
тренерский штаб: гт.Рьянов т.Данчишин т.Нуждин (все из «Чайки») т. И.Знарок («Челмет») т.Шукаев («Саров») сп.дир.Глазов (дебют)
И.Знарок («Челмет») т.Шукаев («Саров») сп.дир.Глазов (дебют)
——————
покинули тренерский штаб: гт.И.Аверкин ст.Н.Воеводин («Чайка»)
игроки остались: в.Мольков в.А.Суханов з.Трубкин з.Н.Полунин з.Огиенко з.В.Шепелев н.Новожилов н.Н.Милёхин н.Н.Томилов (все из «Сарова»)
——————
новые игроки: з.Олендаренко н.Есаян з.Родионычев н.Дон.Стальнов (все из «Торпедо» НН) н.М-м А Михайлов (МХК «Динамо СПб») н.К.Беляев («Рязань») з.Якименко («Торпедо» У-К) н.Смуров («Неман») н.Ведерников («Ижсталь») н.И.Левицкий («Звезда», через ТНН) н.Шураков («Сокол», возврат аренды ТНН) з.П.Ушаков («Тюменский Легион») н.А.Гомоляко («Молот», через «Сочи» и ТНН) з.Павликов («Тамбов», через «Локомотив» и ТНН) з.Лисов («Рубин», через ТНН) в.Доненко («Ценг Тоу») н.В.Михальчук («Принц Георг», WHL, через ТНН) н.М.Цыбин («Ладья») н.Берестенников («Сокол», через «Амур» и ТНН) н.Ден.Морозов («Капитан», через «Локомотив» и ТНН) з.М.Минеев («Динамо» Мск, через ТНН) н.Венгрыжановский («Чайка»)
——————
игроки выбыли: н. Почивалов н.В.Шахворостов (оба в «Торпедо» НН) н.Анд.О.Алексеев («Ростов») з.Парфирьев з.Белоглазов (оба в ЦСК ВВС) з.Анд.С.Миронов н.И.Каштанов (оба в «Рязань») н.Турукин (спорт.директор «Металлурга» Нк) н.Грибов («Ижсталь») н.Крикуненко («Шахтёр» С) н.Бондарук («Торунь») н.Шураков («Лада») н.Д-а Платонов («Чайка») н.Забабурин («ORG») з.Пав.С.Медведев («Сокол», через ТНН и «Амур») н.Каптелин н.Румынин н.А.Полуэктов в.Дм.Безруков з.Белохвостиков
Почивалов н.В.Шахворостов (оба в «Торпедо» НН) н.Анд.О.Алексеев («Ростов») з.Парфирьев з.Белоглазов (оба в ЦСК ВВС) з.Анд.С.Миронов н.И.Каштанов (оба в «Рязань») н.Турукин (спорт.директор «Металлурга» Нк) н.Грибов («Ижсталь») н.Крикуненко («Шахтёр» С) н.Бондарук («Торунь») н.Шураков («Лада») н.Д-а Платонов («Чайка») н.Забабурин («ORG») з.Пав.С.Медведев («Сокол», через ТНН и «Амур») н.Каптелин н.Румынин н.А.Полуэктов в.Дм.Безруков з.Белохвостиков
«Химик» Воскресенск (фарм-клуб «Спартака»)
тренерский штаб: гт.О.Ю.Горбенко («Амур») т.Ал.Ал.Алексеев («Ладья») т.Г.Коротеев т.Д.Коротеев (дебют)
——————
покинули тренерский штаб: гт.Вяч.Козлов («Авангард») т.А.Г.Сырцов («Рязань») т.Вершинин («Красная Армия») т.Привалов
игроки остались: з.Д.Мухин з.Ил.Валеев з.Н-а А Соколов н.Павлюков н.Первов н.Кир.Смирнов н.Баин н.В.Дьяченко н.О.Губин н.Коренев н.Е.Соловьёв н.Котляревский
——————
новые игроки: з.Тришин в.Сковронский н. Тагиров (все из МХК «Спартак») в.А.Трушков з.Ал-й Гришин н.Евг.А.Сорокин н.Г.Шашков н.Д.Черных (все из «Спартака») з.В.Тесленко («Сокол») з.Ил.Давыдов («Ценг Тоу») н.Ил.Карлин («Рубин») н.Зюзякин («Дизель») з.С.Алексеев («Буран») з.Г.Глебов («Зауралье», через «Спартак»)
Тагиров (все из МХК «Спартак») в.А.Трушков з.Ал-й Гришин н.Евг.А.Сорокин н.Г.Шашков н.Д.Черных (все из «Спартака») з.В.Тесленко («Сокол») з.Ил.Давыдов («Ценг Тоу») н.Ил.Карлин («Рубин») н.Зюзякин («Дизель») з.С.Алексеев («Буран») з.Г.Глебов («Зауралье», через «Спартак»)
——————
игроки выбыли: в.С.Большаков («Рязань») н.Е.Кузьменко («Ростов») з.Aл-й Е Kириллов («Металлург» Нк) н.В.Бобров н.Вад.Шутов («Зауралье») з.Гаврилычев («Алматы») з.К.Фомичёв (завершил игровую карьеру) н.Анкудинов («Торос») н.Кляузов («Буран») з.Кобелев («Динамо» Тв) в.Хомченко («Спартак»)
«Хумо» Ташкент, Узбекистан
тренерский штаб: гт.Попихин (НХК, консультант) т.О.Микульчик (экс-«Автомобилист») т.Тортунов («Алматы») гм.Сейейс (ХК «Рига»)
игроки: н.Д.Мошаров («Челмет») н.А-м Воронин з.В.Сорокин (оба из «Спартака») н.Щербина н.Е.Григоренко (оба из «СКА-Невы») н.Ж.Расулов («Комета», ЮХЛ) в.Смирягин («Нефтяник») з.С.Ильин («Торпедо» У-К) н.И.Яценко («Ценг Тоу») з. Пепеляев («Динамо» Мск) з.Таталин («Адмирал») з.Гуляев в.Нечистовский (оба из «Зауралья») з.А.Ерохин («Молот») н.Валуйский («Югра») н.Синявский («Толпар») н.Здунов («Нефтехимик») з.Мережко («Лейтбридж», WHL) в.Р.Голованов («Горняк» У) н.Богацкий («СКА-1946») н.И.Беляков («Амурскиt Тигрs») н.Анд.Ал.Смирнов («Атлант») н.Экк (экс-«Оклахома», WSHL) з.К.Гавриленко (экс-«Октан U18») н.Ворошило («Рязань») н.Пл.Попов («Челмет») з.Ислямов (экс-«Тамбов») н.И.Безруков («Лада»)
Пепеляев («Динамо» Мск) з.Таталин («Адмирал») з.Гуляев в.Нечистовский (оба из «Зауралья») з.А.Ерохин («Молот») н.Валуйский («Югра») н.Синявский («Толпар») н.Здунов («Нефтехимик») з.Мережко («Лейтбридж», WHL) в.Р.Голованов («Горняк» У) н.Богацкий («СКА-1946») н.И.Беляков («Амурскиt Тигрs») н.Анд.Ал.Смирнов («Атлант») н.Экк (экс-«Оклахома», WSHL) з.К.Гавриленко (экс-«Октан U18») н.Ворошило («Рязань») н.Пл.Попов («Челмет») з.Ислямов (экс-«Тамбов») н.И.Безруков («Лада»)
«Ценг Тоу» Цзилинь, China
тренерский штаб: гт.Р.Гусманов («Челмет») т.Д.Андреев т.Тан гм.ЧжоПу
——————
покинули тренерский штаб: гт.Кирдяшов
игроки остались: з.М-м Иванов н.Ег.С.Кожевников н.А.Швецов н.Гайсин н.Чемерикин н.Ионин н.Ал-р Ю Васильев н.М.Зарипов н.Епимахин
——————
новые игроки: н.Ибраев («Астана») н.Сейтанов («Номад») в.Гайдуллин («Горняк» У) з.Джангарашев («Бингхэмтон Джуниор Сенаторз», NA3HL) з.Дэх.Чжан н.Х. У (оба из «КРС-ORG») з.Вахрутдинов («Мамонты Югры») з.А.Рыжих (ЦСК ВВС) з.В.Лысенко («Молот») з.Курдюков («Ижсталь») з.Жигалов («Южный Урал-Металлург») н.Дм.Гусманов (экс-«Южный Урал-Металлург») з.Туспеков («Горняк» Рд) в.Красановски (МХК «КРС Хэйлунцзян») н.Руз.Галеев («Алматы») в.Т.Чжоу (экс-студ.сб.China) н.У.Гильманов з.В.Арбузов (оба из «Нефтяника») н.Ег.В.Иванов («Югра») з.Б.Гендунов («Амурские Тигры») в.Д-л Яковлев (ЦСК ВВС)
У (оба из «КРС-ORG») з.Вахрутдинов («Мамонты Югры») з.А.Рыжих (ЦСК ВВС) з.В.Лысенко («Молот») з.Курдюков («Ижсталь») з.Жигалов («Южный Урал-Металлург») н.Дм.Гусманов (экс-«Южный Урал-Металлург») з.Туспеков («Горняк» Рд) в.Красановски (МХК «КРС Хэйлунцзян») н.Руз.Галеев («Алматы») в.Т.Чжоу (экс-студ.сб.China) н.У.Гильманов з.В.Арбузов (оба из «Нефтяника») н.Ег.В.Иванов («Югра») з.Б.Гендунов («Амурские Тигры») в.Д-л Яковлев (ЦСК ВВС)
——————
игроки выбыли: н.Н.Лукин («Лада») з.Ушнев («Динамо» СПб) з.Д.Громов в.Перевозчиков з.В.Садиков (все в «Торпедо» У-К) з.И.Давыдов («Химик») з.Е.Калинин («Молот») н.И.Яценко («Хумо») з.Е.Дубовицкий («Ермак») н.Ег.Лебедев («Сокол», через «Югру») н.Шибаев («Днепр Херсон») з.Анд.Н.Алексеев («Рубин») з.Раров («Тайфун») в.Доненко («Торпедо-Горький») з.Ш.Ван («КРС-БСУ») в.Хоу н.Ант.Крысанов
ЦСК ВВС Самара (фарм-соглашение с «Нефтехимиком»)
тренерский штаб: гт.Ал.Ю.Соколов т.О.Юшин («Челны») т.И.Мисбахов т. Н.Скатов (оба завершили игровую карьеру) трен-конс.Шарнин
Н.Скатов (оба завершили игровую карьеру) трен-конс.Шарнин
——————
покинули тренерский штаб: т.С.Шиханов т.Булдаков сп.дир.В.Шиханов
игроки остались: з.М-м А Гусев н.Набиуллин (оба в аренде от НХ) н.Сыромятников н.Е.Грошев (оба в аренде от «Лады») н.А.Степаненко н.Юнгов н.Д.Гареев
——————
новые игроки: н.Г.Куликов («Горняк» Рудный) н.Л.Николенко («Ладья») н.Рукин з.П.П.Десятков (оба из «Рязани») в.Мамедов («Дин.Харьков») н.Н.Свинцицкий («Тамбов») з.Парфирьев з.Белоглазов (оба из «Сарова») н.Тимиров («Барс», аренда) з.Ант.Кудрявцев з.Ананьин (оба из «Темиртау») з.Максюков («Юниор») н.А.Голубович з.Пузанов (оба из «Реактора») з.Ег.Сорокин («Молот») н.Чередниченко («Комета-U18») в.Попков н.Колычев н.Ахметзянов н.Юнышев (все из «Мордовии») н.Д.Каштанов («Торпедо» У-К) з.Ден.Кузьмин («Амур») з.Г.Семёнов н.А.Хисамутдинов в.Ф.Коротаев з.Ал.Волгин (все из «Нефтехимика») н.С.Скворцов («Пинские Ястребы»)
——————
игроки выбыли: н.Разумняк («Номад») з. Шкибтан (МХК «Динамо СПб») н.В.Журавлёв з.Рин.Шафигуллин з.Т.Булатов (все в «Ладу») н.Мнихович («Рубин») н.Лудцев («Металлург» Нк) н.Мингачёв («Красная Армия») з.А.Рыжих («Ценг Тоу») в.Жучин («Чебоксары») н.И.Угольников («Лада», через «Нефтехимик») н.Лисицын (МХК «Кристалл») з.Н.Манухов н.Литвяк з.Пангелов-Юлдашев н.Д.Тесленко («Актобе») в.Д-л Яковлев («Ценг Тоу») з.Сенников («Оренбург»)
Шкибтан (МХК «Динамо СПб») н.В.Журавлёв з.Рин.Шафигуллин з.Т.Булатов (все в «Ладу») н.Мнихович («Рубин») н.Лудцев («Металлург» Нк) н.Мингачёв («Красная Армия») з.А.Рыжих («Ценг Тоу») в.Жучин («Чебоксары») н.И.Угольников («Лада», через «Нефтехимик») н.Лисицын (МХК «Кристалл») з.Н.Манухов н.Литвяк з.Пангелов-Юлдашев н.Д.Тесленко («Актобе») в.Д-л Яковлев («Ценг Тоу») з.Сенников («Оренбург»)
«Челмет» Челябинск (фарм-клуб «Трактора»)
тренерский штаб: гт.Смельницкий («Белые Медведи») т.С.Тертышный («Металлург» Жл) т.Татаринцев (СДЮСШОР «Трактор») т.Штрахов («Трактор»)
——————
покинули тренерский штаб: гл.И.Знарок («Торпедо-2») т.Вал.Никулин т.Р.Гусманов («Ценг Тоу») т.Вик.Демченко
игроки остались: в.Подскребалин в.С.Мыльников з.Л.Лавриненко з.А.Шепелев з.Мокрушев з.Шурыгин з.Складчиков н.Кориневский н.Кошурников н.Арк.Шафигулин н.И.Зиновьев н.Шадрин н.М.Коновалов н.Дм.К.Тарасов н.Н.Тертышный н.Подкорытов н.Е.Степанов н.Ширяев
——————
новые игроки: в. Д.Шаров з.И.Гатиятуллин з.Кирпичников н.Кулиев н.Д.Мансуров (все из «Белых Медведей») з.Назаркин («Динамо» СПб, через ТЧ) з.Микулович («Каламазу», ECHL через ТЧ) в.Сухачёв з.Дм.Вит.Алексеев з.Иг.Исаев з.А-м Бородкин н.Aл-р П Шаров (все из «Трактора»)
Д.Шаров з.И.Гатиятуллин з.Кирпичников н.Кулиев н.Д.Мансуров (все из «Белых Медведей») з.Назаркин («Динамо» СПб, через ТЧ) з.Микулович («Каламазу», ECHL через ТЧ) в.Сухачёв з.Дм.Вит.Алексеев з.Иг.Исаев з.А-м Бородкин н.Aл-р П Шаров (все из «Трактора»)
——————
игроки выбыли: н.Д.Мошаров («Хумо») з.Шушарин («Красноярские Рыси») н.Уфимцев («Южный Урал») з.Жульдиков («Торос», через «С.Юлаев») з.Петриков («Рубин», через аренду от «Трактора») н.Афонасьевский («Звезда», через ЦСКА) з.Мякишев в.Вик.Селивёрстов (оба в «Белые Медведи») н.Пл.Попов («Хумо») з.Костарев («Номад») н.Ю.Могильников («Химик» Нвп) н.Кобяков («Темиртау») н.А-м Мелихов («Рязань») з.С.Лукин в.Е.Угрюмов в.К.М.Кузьмин н.Сентюрин з.Вад.Никулин
«Югра» Ханты-Мансийск
тренерский штаб: гт.Епанчинцев (экс-«Спартак») т.Костючёнок («Сарыарка») т.Е.Фёдоров т.Яцкевич (экс-«Сарыарка») т.Галимжанов гм.С.Гусев
——————
покинули тренерский штаб: гт.А.Ждахин т.Бульин (оба в «Торпедо» У-К) т. С.Храмцов
С.Храмцов
игроки остались: в.Е.Иванников в.К.Волков (аренда от СКА) з.Чуркин з.Воропаев з.Валентенко н.А.Митрофанов н.Р.Бердников н.Ал-р Вл Беляев н.И.Фищенко н.Берлёв н.М-м Аскаров н.Пилипенко (аренда от «Автомобилиста»)
——————
новые игроки: н.Арсенюк н.Борисенков (оба из «Горняка» Уч) з.М.Рогов («Торос») з.Верёвкин («Рубин») н.Зиазов н.Махановский н.Ряшенцев (все из «Сарыарки») н.Раисов («Динамо» Мск) н.Латышевич з.С.Телегин н.Чусовитин (все из «Мамонтов Югры») з.В.Мирошниченко (НХК) з.Дм.А.Сергеев («Алматы») з.Лекомцев («Северсталь») н.А.Акмальдинов («Сочи») н.Сигарёв («Лада»)
——————
игроки выбыли: в.Шилин («Сокол») н.Рожковский н.Ант.Ковалёв н.Н.Самохвалов (все в «Ижсталь» через «Авангард») н.Грымзин з.Катин (оба в «Горняк» Уч) з.Э.Шевченко з.Д.Галеев (оба в «Металлург» Нк) н.Валуйский («Хумо») н.К.Кожевников («Ермак», через «Адмирал») з.С.А.Кузнецов н.Дм.Э.Михайлов н.Голоднюк (все в «Сарыарку») н.Матерухин («Могилёв») н.Евг.Ал.Орлов («Ижсталь») н. Рычагов («Динамо» СПб) н.Игнашин («Торпедо» У-К) з.Д.Д.Юшкевич («Звезда») н.Кир.Попов («Мамонты Югры») н.Ег.В.Иванов («Ценг Тоу») з.Р.Рачинский з.Г.Ващенко н.Вит.Вит.Карамнов з.Дан.Стальнов
Рычагов («Динамо» СПб) н.Игнашин («Торпедо» У-К) з.Д.Д.Юшкевич («Звезда») н.Кир.Попов («Мамонты Югры») н.Ег.В.Иванов («Ценг Тоу») з.Р.Рачинский з.Г.Ващенко н.Вит.Вит.Карамнов з.Дан.Стальнов
«Южный Урал» Орск
тренерский штаб: гт.О.Черкасов т.Музычко т.Скомороха («Сарматы») т.Бучельников
игроки остались: з.А-м Чистяков з.Ванин з.Н.Жилин з.Е.Бардаков з.Жлоба н.Е.Дорофеев (оба в аренде от «Металлурга» Мг) н.Марченков н.Ил.Д.Иванов н.Марзоев н.Ден.Кондратьев н.И.Скворцов
——————
новые игроки: в.Кислицын («Липтовски Микулаш») з.Арс.Нургалиев н.Д.Антонов (оба из «Горняка» Уч) з.Ал-й Кожевников («Рубин») з.А.Смолин н.Хасаншин (оба из «Бурана») н.Ал-й Прохоров («Ростов») в.Филоненко («Нефтяник», через «Звезду») н.Курепанов («Зауралье») н.Уфимцев («Челмет») н.Шипин («Трактор») н.Мурзин («Буран») н.Тимашев («Сарматы») н.Бархаткин («Спутник» А) з.Д.Егоров (МХК «Динамо» Мс)
——————
игроки выбыли: з.Куршук («Иртыш») н. Г.Пивунов (МХК «Динамо СПб») в.Синягин з.И.Гавриленко (оба в «Динамо» СПб) з.В.Репин («Тамбов») н.Сметанин н.Ден.Фахрутдинов н.Д.Скатов (все в «Молот») н.Чугуев («Рубин») н.Кудрин («Рязань») н.И.Черкасов («Актобе») з.П.Сахаров («Сарматы») з.Рябоконов («Металлург» Нк) н.Соколка («Юниор») з.Крупенко н.Ст.Соловьёв в.Д.Перетягин н.А.Севанькаев
Г.Пивунов (МХК «Динамо СПб») в.Синягин з.И.Гавриленко (оба в «Динамо» СПб) з.В.Репин («Тамбов») н.Сметанин н.Ден.Фахрутдинов н.Д.Скатов (все в «Молот») н.Чугуев («Рубин») н.Кудрин («Рязань») н.И.Черкасов («Актобе») з.П.Сахаров («Сарматы») з.Рябоконов («Металлург» Нк) н.Соколка («Юниор») з.Крупенко н.Ст.Соловьёв в.Д.Перетягин н.А.Севанькаев
Переходы в «Спартаке» 2020
В связи с большим количеством заказов отправка посылок перед новогодними праздниками может задержаться на 1-2 дня.
Последний день отправки заказов 30 декабря, все заказы оформленные после 29 декабря будут отправляться 3 января и 6 января 2022 года. В стандартном режиме отправки возобновятся с 10 января.
| ПРИШЛИ | УШЛИ | ОСТАЛИСЬ |
| н.Данила Квартальнов** | в. Юлиус Гудачек Юлиус Гудачек | н.Евгений Сорокин ** |
| в.Алексей Красиков** | в.Никита Беспалов | з.Аким Тришин ** |
| з.Том Хеед | н.Лукаш Радил | н.Дмитрий Силантьев ** |
| з.Дмитрий Костенко | н.Робин Ганзл | в.Андрей Сковронский** |
| з.Степан Фролов | н.Илья Зубов | н.Егор Круженков** |
| з.Никита Громов** | з.Евгений Кулик | |
| з.Семён Ручкин** | з.Андрей Кутейкин | |
| н.Илья Баранов** | н. Йори Лехтеря Йори Лехтеря | |
| н.Вадим Фаттахов** | н.Анатолий Никонцев | |
| н.Валентин Демченко** | з.Алексей Гришин | |
| н.Василий Пономарёв** | н.Илья Аркалов | |
| з.Максим Афанасьев** | н.Александр Торченюк | |
| | н.Артём Фёдоров | |
| | н.Павел Чернов | |
| | н.Михаил Юньков | |
| | н.Павел Медведев | |
| | н. Илья Кляузов Илья Кляузов | |
| | н.Богдан Львутин | |
| | з.Антон Мишаков | |
| | з.Дмитрий Александров | |
| | з.Илья Селин | |
| | н.Мартин Бакош | |
| | в.Александр Трушков | |
| | з.Юрий Козловский | |
| | н.Антон Первов | |
ОСА — ограниченно свободный агент
**- двухсторонний контракт (КХЛ / ВХЛ)
***- трёхсторонний контракт (КХЛ / ВХЛ / МХЛ)
Будь в курсе всех новостей, подпишись!
Обещаем присылать только самые важные новости, никакого спама!
Подписаться Введите адрес электронной почты
Самат Данияр (спортсмен, хоккей, Казахстан): новости, биография, статистика спортсмена
Самат Данияр (спортсмен, хоккей, Казахстан): новости, биография, статистика спортсмена | Vesti. kz gR10mvn2iuHu8m7V00q4osey2lwymbhkBAZ0pRUV
Войти через социальную сеть
kz gR10mvn2iuHu8m7V00q4osey2lwymbhkBAZ0pRUV
Войти через социальную сетьПожалуйста, подождите… Укажите email Укажите имя или псевдоним Укажите пароль Для регистрации на сайте Вы должны принять Правила сообщества Для редактирования профиля необходимо авторизоваться на сайте Укажите корректный Email material_dobavlen_v_izbrannoe Добавить в избранное Убрать из избранного Пароли не совпадают Задайте пароль для входа на сайт Хороший пароль должен содержать строчные, заглавные латинские буквы и цифры. Рекомендуется добавлять знаки препинания и задавать длину пароля не менее 8 символов Спасибо за Ваш голос! Добавить +1 Убрать +1 Выберите вариант ответа
Место
Вход на сайт
Войти через социальную сеть:>
 org/PostalAddress»>
org/PostalAddress»>Барыс зщ 65
Проектирование и анализ модели сети Петри сети взаимодействия фон Хиппеля-Линдау (VHL) с супрессором опухоли Анализ модели сети Петри сети взаимодействия фон Хиппеля-Линдау (VHL) с опухолевым супрессором.
 ПЛОС ОДИН 9(6):
е96986.
https://doi.org/10.1371/journal.pone.0096986
ПЛОС ОДИН 9(6):
е96986.
https://doi.org/10.1371/journal.pone.0096986Редактор: Manuela Helmer-Citterich, Римский университет Тор Вергата, Италия
Поступила в редакцию: 4 сентября 2013 г.; Принято: 14 апреля 2014 г .; Опубликовано: 2 июня 2014 г.
Авторские права: © 2014 Minervini et al.Это статья с открытым доступом, распространяемая в соответствии с лицензией Creative Commons Attribution License, которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии указания оригинального автора и источника.
Финансирование: Эта работа была поддержана Associazione Italiana per la Ricerca sul Cancro (AIRC) Грант MFAG 12740 для ST. GM является научным сотрудником AIRC (http://www.airc.it/). Спонсоры не участвовали в разработке исследования, сборе и анализе данных, принятии решения о публикации или подготовке рукописи.
Конкурирующие интересы: Авторы также хотели бы заявить, что: соавтор Сильвио Тосатто является членом редакционной коллегии PLOS ONE. Это не меняет приверженности авторов всем политикам PLOS ONE в отношении обмена данными и материалами.
Введение
Патологическая дерегуляция клеточных путей часто приводит к семейству сложных и взаимосвязанных заболеваний, обычно называемых раком [1]. Рак является многофакторным заболеванием, в развитии которого участвуют различные причины.Было разработано несколько вычислительных методов для изучения функциональных путей, участвующих в онкогенезе. Некоторые из них сосредоточены на дифференциальной экспрессии генов между здоровыми и патологическими тканями [2]–[3], на анализе сетей белок-белковых взаимодействий [4]–[5] или на моделировании молекулярной динамики [6]. Другие методы приближаются к заболеванию путем дискретизации патологических компонентов, которые приводят к опухоли [7]. Все эти подходы очень эффективны, когда переменные, связанные с болезнью, хотя и сложны, но хорошо известны и изучены. К многофакторному заболеванию можно подойти с помощью математической теории, построив теоретическую модель, в которой компоненты клетки связаны друг с другом. В биологии несколько задач касались теории сетей [8]–[9]. Сеть — это группа объектов, сильно связанных друг с другом (например, белки и ферменты пути или животные, принадлежащие к взаимодействующим популяциям). Их построение и последующее моделирование осуществляется путем математического анализа связей между найденными в системе узлами и их поведения во времени [10].Биологическая сеть обычно состоит из белков, нуклеиновых кислот и кофакторов, связанных биологическими реакциями, такими как образование белковых комплексов или регуляция активности ферментов [10]. Синдром фон Хиппеля-Линдау (VHL) [11] является хорошим примером для изучения сетевой теории, применяемой к раку, из-за схожей истории болезни и патологического фенотипа, которые разделяют пациенты. В то время как наследственные виды рака составляют лишь небольшую часть всех опухолей человека, их исследование представляет собой проблему для понимания пути, ведущего к образованию опухоли.
К многофакторному заболеванию можно подойти с помощью математической теории, построив теоретическую модель, в которой компоненты клетки связаны друг с другом. В биологии несколько задач касались теории сетей [8]–[9]. Сеть — это группа объектов, сильно связанных друг с другом (например, белки и ферменты пути или животные, принадлежащие к взаимодействующим популяциям). Их построение и последующее моделирование осуществляется путем математического анализа связей между найденными в системе узлами и их поведения во времени [10].Биологическая сеть обычно состоит из белков, нуклеиновых кислот и кофакторов, связанных биологическими реакциями, такими как образование белковых комплексов или регуляция активности ферментов [10]. Синдром фон Хиппеля-Линдау (VHL) [11] является хорошим примером для изучения сетевой теории, применяемой к раку, из-за схожей истории болезни и патологического фенотипа, которые разделяют пациенты. В то время как наследственные виды рака составляют лишь небольшую часть всех опухолей человека, их исследование представляет собой проблему для понимания пути, ведущего к образованию опухоли. В 2010 году Хайнер и соавт. впервые подошел к VHL, используя так называемые сети моделирования Petri Net (PN) [12]. Их работа, вдохновленная предыдущей теоретической моделью клеточных путей, связанных с кислородом [13] [14], была предварительным исследованием основной системы восприятия кислорода и ее связи с началом VHL. Хайнер и его коллеги предложили три разных функциональных модуля, ответственных за контроль сети гипоксии и за деградацию HIF-1α [12]. Другими словами, они предположили, что наследственные формы рака, такие как различные проявления VHL, являются результатом различных и одновременно нарушенных метаболических путей.
В 2010 году Хайнер и соавт. впервые подошел к VHL, используя так называемые сети моделирования Petri Net (PN) [12]. Их работа, вдохновленная предыдущей теоретической моделью клеточных путей, связанных с кислородом [13] [14], была предварительным исследованием основной системы восприятия кислорода и ее связи с началом VHL. Хайнер и его коллеги предложили три разных функциональных модуля, ответственных за контроль сети гипоксии и за деградацию HIF-1α [12]. Другими словами, они предположили, что наследственные формы рака, такие как различные проявления VHL, являются результатом различных и одновременно нарушенных метаболических путей.
Болезнь Гиппеля-Линдау
Белок фон Хиппеля-Линдау (pVHL) является продуктом гена фон Хиппеля-Линдау, расположенного в коротком плече 3-й хромосомы и постоянно транскрибируемого как в эмбриональных, так и во взрослых тканях [15]. Мутации pVHL связаны с патологическим исходом, называемым синдромом VHL, наследственной формой рака [16]. Синдром VHL характеризуется кистами и опухолями, растущими в определенных частях организма [16]–[17]. Это считается тяжелым аутосомно-доминантным генетическим заболеванием с наследованием одного человека из более чем 35 000 [18].Опухолевые поражения, которые могут быть как доброкачественными, так и злокачественными, обычно локализуются в сетчатке, надпочечниках, придатках яичек, центральной нервной системе, почках и поджелудочной железе [19]. Как генетическое заболевание, синдром VHL следует принципу двух ударов Кнудсона. Копия гена мутирует в зародышевой линии, но другая копия гена по-прежнему производит функциональный белок. Полная инактивация белка возникает в течение жизни за счет соматической инактивации оставшейся функциональной копии [20]. Наоборот, мутации, возникающие в период раннего формирования плода, приводят к неудачному развитию [21].Ген pVHL имеет 11 213 пар оснований, включая три экзона [18], а конечный транскрипт представляет собой белок, обычно присутствующий в двух изоформах: pVHL30 и pVHL19, состоящих из 213 и 160 остатков соответственно. Ни одна из изоформ не содержит известного ферментативного домена, а скорее служит многоцелевым адапторным белком, участвующим во множественных белок-белковых взаимодействиях [22].
Это считается тяжелым аутосомно-доминантным генетическим заболеванием с наследованием одного человека из более чем 35 000 [18].Опухолевые поражения, которые могут быть как доброкачественными, так и злокачественными, обычно локализуются в сетчатке, надпочечниках, придатках яичек, центральной нервной системе, почках и поджелудочной железе [19]. Как генетическое заболевание, синдром VHL следует принципу двух ударов Кнудсона. Копия гена мутирует в зародышевой линии, но другая копия гена по-прежнему производит функциональный белок. Полная инактивация белка возникает в течение жизни за счет соматической инактивации оставшейся функциональной копии [20]. Наоборот, мутации, возникающие в период раннего формирования плода, приводят к неудачному развитию [21].Ген pVHL имеет 11 213 пар оснований, включая три экзона [18], а конечный транскрипт представляет собой белок, обычно присутствующий в двух изоформах: pVHL30 и pVHL19, состоящих из 213 и 160 остатков соответственно. Ни одна из изоформ не содержит известного ферментативного домена, а скорее служит многоцелевым адапторным белком, участвующим во множественных белок-белковых взаимодействиях [22].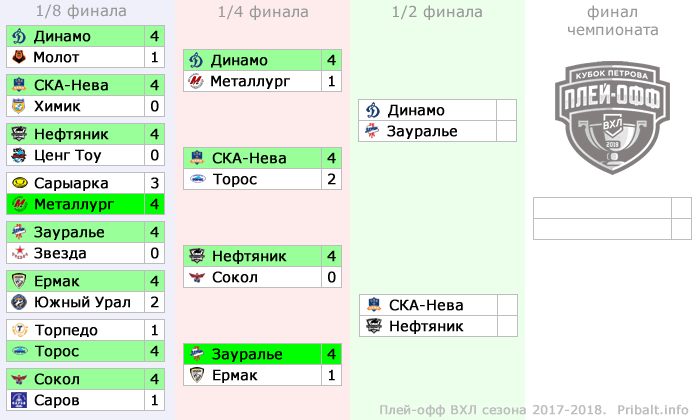 Структура pVHL организована в α- и β-доменах, и было показано, что ее стабильность обеспечивается прямым взаимодействием с другими белками, такими как элонгины B и C [23].И элонгин B, и C также необходимы для наиболее охарактеризованной функции pVHL, зависимой от убиквитинирования деградации Hypoxia Inducible Factor (HIF) через протеасомы [24]. Однако pVHL считается многоцелевым белком из-за большого количества известных взаимодействующих факторов. На момент написания в базе данных IntAct [25] было представлено более 200 различных партнеров по взаимодействию, причем некоторые из них конкурируют за один и тот же сайт связывания элонгина. Действительно, pVHL был обнаружен в различных клеточных компартментах и, по-видимому, участвует во многих различных клеточных процессах, таких как апоптоз, клеточная пролиферация, выживание и подвижность [26].Учитывая огромное количество интеракторов и множество клеточных локализаций, было описано или выдвинуто предположение о многих различных функциях, таких как регуляция цитоплазматических микротрубочек во время митоза [27] и отложение эндотелиального внеклеточного матрикса [28].
Структура pVHL организована в α- и β-доменах, и было показано, что ее стабильность обеспечивается прямым взаимодействием с другими белками, такими как элонгины B и C [23].И элонгин B, и C также необходимы для наиболее охарактеризованной функции pVHL, зависимой от убиквитинирования деградации Hypoxia Inducible Factor (HIF) через протеасомы [24]. Однако pVHL считается многоцелевым белком из-за большого количества известных взаимодействующих факторов. На момент написания в базе данных IntAct [25] было представлено более 200 различных партнеров по взаимодействию, причем некоторые из них конкурируют за один и тот же сайт связывания элонгина. Действительно, pVHL был обнаружен в различных клеточных компартментах и, по-видимому, участвует во многих различных клеточных процессах, таких как апоптоз, клеточная пролиферация, выживание и подвижность [26].Учитывая огромное количество интеракторов и множество клеточных локализаций, было описано или выдвинуто предположение о многих различных функциях, таких как регуляция цитоплазматических микротрубочек во время митоза [27] и отложение эндотелиального внеклеточного матрикса [28]. С другой стороны, учитывая огромное количество игроков, вовлеченных в синдром VHL, и отсутствие надежных кинетических данных, подход, основанный на PN, может быть предпочтительным вариантом для моделирования всего пути VHL.
С другой стороны, учитывая огромное количество игроков, вовлеченных в синдром VHL, и отсутствие надежных кинетических данных, подход, основанный на PN, может быть предпочтительным вариантом для моделирования всего пути VHL.
Сеть Петри для путей взаимодействия
С момента их изобретения Карлом Адамом Петри в начале 60-х годов СП в основном использовались для описания технических систем, но позже также была продемонстрирована полезность для описания биологических и биохимических функций [29].PN были успешно использованы во многих исследованиях для описания биологических сетей [30], таких как регуляция и этиопатология при мышечной дистрофии Дюшенна человека [31] и сеть ответа на гипоксию [12]. СП представляют собой качественные математические модели, которые могут графически представлять многие типы объектов, не только метаболиты, но и различные состояния белков, и полезны для моделирования сетей, в которых задействованы не только метаболиты. Действительно, PN могут быть мощным инструментом для изучения всех одновременных взаимодействий в определенном пути, даже если белки или кинетика недостаточно известны.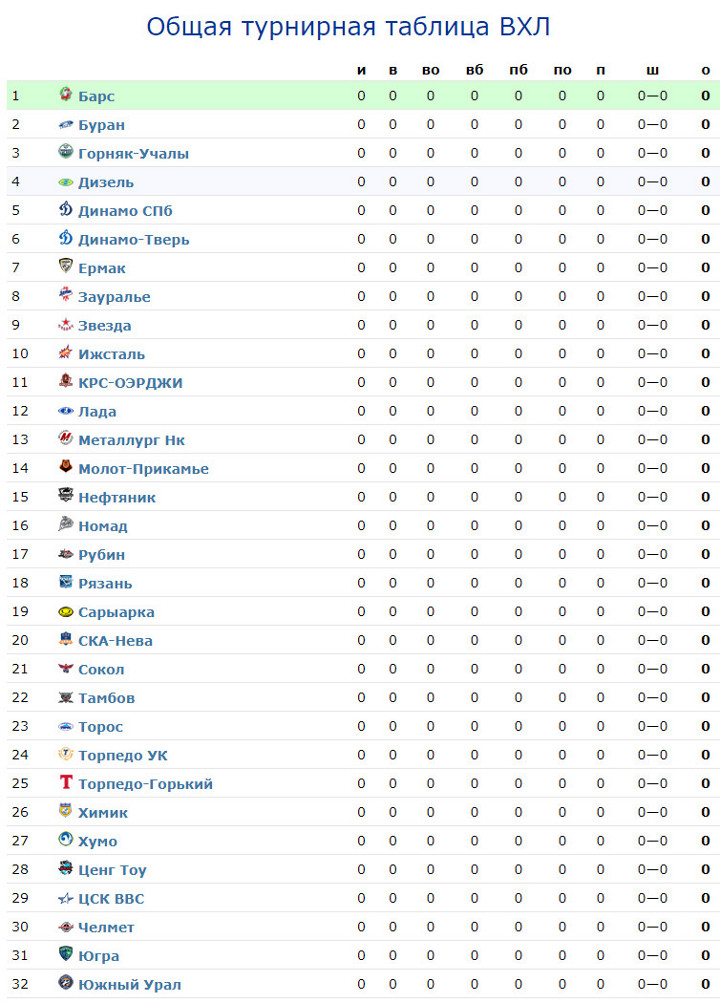 Из-за большого количества различных функций pVHL, участвующих в прогрессировании заболевания VHL, мы решили расширить анализ на основе PN [12], увеличив количество рассматриваемых белок-белковых взаимодействий. Мы создали новую вручную созданную модель PN всей системы регуляции VHL, собрав данные из литературы и включив сигнальные пути и глюкозный метаболизм. Чтобы построить реалистичную сеть, в качестве источника использовалась литература как по биохимическим экспериментам, так и по предсказаниям in silico .Было решено построить PN только с подтвержденными взаимодействиями pVHL, функция которых также была известна. Полученное PN было подтверждено с помощью анализа конкретных свойств, как было предложено в предыдущих исследованиях с использованием того же метода [29]. После проверки структуры PN было выполнено in silico нокаутов конкретных белков, чтобы наблюдать различное поведение сети и получающийся в результате биологический эффект.
Из-за большого количества различных функций pVHL, участвующих в прогрессировании заболевания VHL, мы решили расширить анализ на основе PN [12], увеличив количество рассматриваемых белок-белковых взаимодействий. Мы создали новую вручную созданную модель PN всей системы регуляции VHL, собрав данные из литературы и включив сигнальные пути и глюкозный метаболизм. Чтобы построить реалистичную сеть, в качестве источника использовалась литература как по биохимическим экспериментам, так и по предсказаниям in silico .Было решено построить PN только с подтвержденными взаимодействиями pVHL, функция которых также была известна. Полученное PN было подтверждено с помощью анализа конкретных свойств, как было предложено в предыдущих исследованиях с использованием того же метода [29]. После проверки структуры PN было выполнено in silico нокаутов конкретных белков, чтобы наблюдать различное поведение сети и получающийся в результате биологический эффект.
Методы
Сеть была разработана в рамках Snoopy PN (версия 2, ревизия 1. 13) [32], соблюдая математический формализм PN, описанный в [12]–[33]. Было показано, что СП полезны для описания дискретных и параллельных процессов в простом графическом представлении [30] и использовались для описания биомедицинских процессов благодаря их способности представлять последовательные этапы процесса. Методы моделирования СП активно используются для описания, моделирования, анализа и прогнозирования поведения биологических систем. Среда Snoopy PN предоставляет расширяемую мультиплатформенную среду для проектирования, анимации и моделирования сетей Петри [32].Мы выбрали Snoopy для облегчения будущих расширений пути VHL, представленного здесь. Среди различных доступных типов PN был выбран стандартный PN, чтобы ограничить количество переменных. Для анализа и проверки PN использовались анализаторы Charlie и PInA (34). Кроме того, для проверки биологической надежности модели использовали эксперименты с нокаутом in silico . Проверка структурной модели проводилась путем анализа Т-инвариантов, чтобы продемонстрировать, покрыта ли система Т-инвариантами, и подтвердить биологический смысл каждого инварианта.
13) [32], соблюдая математический формализм PN, описанный в [12]–[33]. Было показано, что СП полезны для описания дискретных и параллельных процессов в простом графическом представлении [30] и использовались для описания биомедицинских процессов благодаря их способности представлять последовательные этапы процесса. Методы моделирования СП активно используются для описания, моделирования, анализа и прогнозирования поведения биологических систем. Среда Snoopy PN предоставляет расширяемую мультиплатформенную среду для проектирования, анимации и моделирования сетей Петри [32].Мы выбрали Snoopy для облегчения будущих расширений пути VHL, представленного здесь. Среди различных доступных типов PN был выбран стандартный PN, чтобы ограничить количество переменных. Для анализа и проверки PN использовались анализаторы Charlie и PInA (34). Кроме того, для проверки биологической надежности модели использовали эксперименты с нокаутом in silico . Проверка структурной модели проводилась путем анализа Т-инвариантов, чтобы продемонстрировать, покрыта ли система Т-инвариантами, и подтвердить биологический смысл каждого инварианта. Использование Т- и Р-инвариантов обусловлено их собственными свойствами: они представляют собой набор (соответственно переходов или мест), позволяющих воспроизвести одно и то же состояние после n преобразований. P-инвариант представляет собой набор мест, в которых количество токенов постоянно и не зависит от скорости срабатывания. Вместо этого T-инвариант представляет набор переходов, который циклически возвращается, чтобы показать тот же начальный набор. Биологически инвариант P может представлять процесс регуляции белка, тогда как инвариант T может представлять собой циклические биохимические преобразования, такие как метаболические реакции.С этой целью вычисленные инварианты были сгруппированы в максимальные общие наборы переходов (MCTS) и кластеры, первые из которых основаны на наличии определенных наборов переходов внутри различных Т-инвариантов, а вторые — на сходстве между Т-инвариантами. В зависимости от результирующей квадратной матрицы будут определены различные количества кластеров.
Использование Т- и Р-инвариантов обусловлено их собственными свойствами: они представляют собой набор (соответственно переходов или мест), позволяющих воспроизвести одно и то же состояние после n преобразований. P-инвариант представляет собой набор мест, в которых количество токенов постоянно и не зависит от скорости срабатывания. Вместо этого T-инвариант представляет набор переходов, который циклически возвращается, чтобы показать тот же начальный набор. Биологически инвариант P может представлять процесс регуляции белка, тогда как инвариант T может представлять собой циклические биохимические преобразования, такие как метаболические реакции.С этой целью вычисленные инварианты были сгруппированы в максимальные общие наборы переходов (MCTS) и кластеры, первые из которых основаны на наличии определенных наборов переходов внутри различных Т-инвариантов, а вторые — на сходстве между Т-инвариантами. В зависимости от результирующей квадратной матрицы будут определены различные количества кластеров.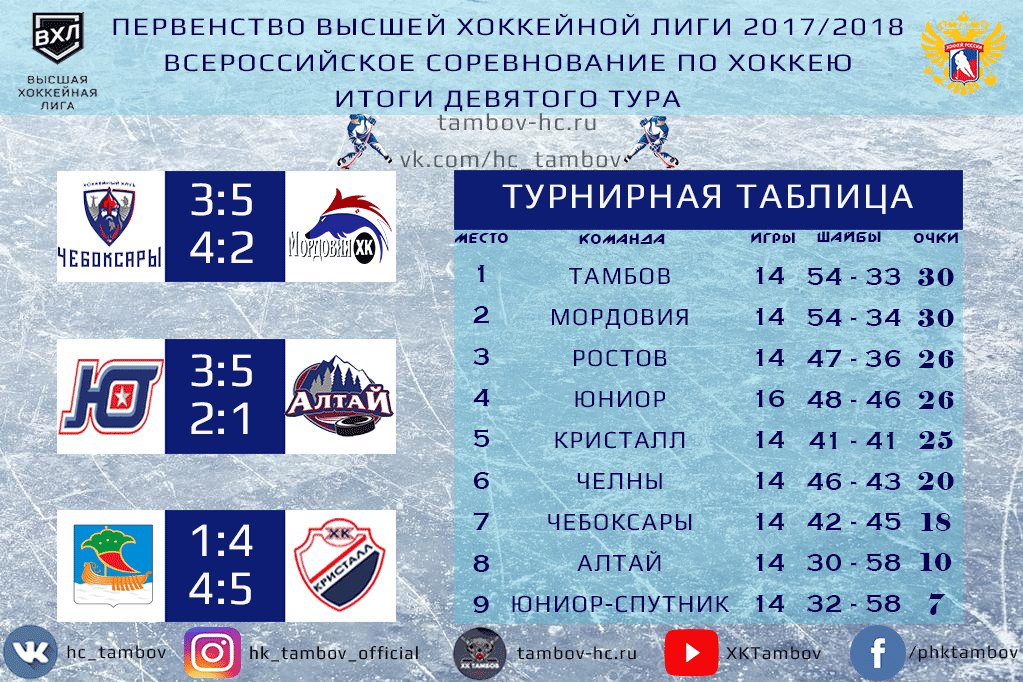 Там, где MCTS создают дизъюнктивные сети, кластеры объединяют похожие T-инварианты. Поведенческая валидация проводилась путем выборочного удаления токенов внутри модели, имитируя возможные биологические нарушения, такие как мутации, вызывающие заболевание.Полученное поведение сети сравнивали с тем, что сообщается в литературе. Общее время выполнения для вычисления инвариантов составило менее десяти секунд на основной рабочей станции Linux x86. Источники литературы, использованные для построения модели, указаны в таблице S1. Среда Snoopy для построения PN, инструменты Charlie и PInA для анализа доступны на веб-сайте (URL: http://www-dssz.informatik.tu-cottbus.de/DSSZ/Software). Наконец, модель использовалась для имитации поведения сети посредством визуального контроля как движения токенов, так и их накопления в определенных частях сети.Для визуального объяснения движения токена в PN см. Видео S1.
Там, где MCTS создают дизъюнктивные сети, кластеры объединяют похожие T-инварианты. Поведенческая валидация проводилась путем выборочного удаления токенов внутри модели, имитируя возможные биологические нарушения, такие как мутации, вызывающие заболевание.Полученное поведение сети сравнивали с тем, что сообщается в литературе. Общее время выполнения для вычисления инвариантов составило менее десяти секунд на основной рабочей станции Linux x86. Источники литературы, использованные для построения модели, указаны в таблице S1. Среда Snoopy для построения PN, инструменты Charlie и PInA для анализа доступны на веб-сайте (URL: http://www-dssz.informatik.tu-cottbus.de/DSSZ/Software). Наконец, модель использовалась для имитации поведения сети посредством визуального контроля как движения токенов, так и их накопления в определенных частях сети.Для визуального объяснения движения токена в PN см. Видео S1.
Наличие модели
Результирующая модель PN болезни VHL доступна в файле S1.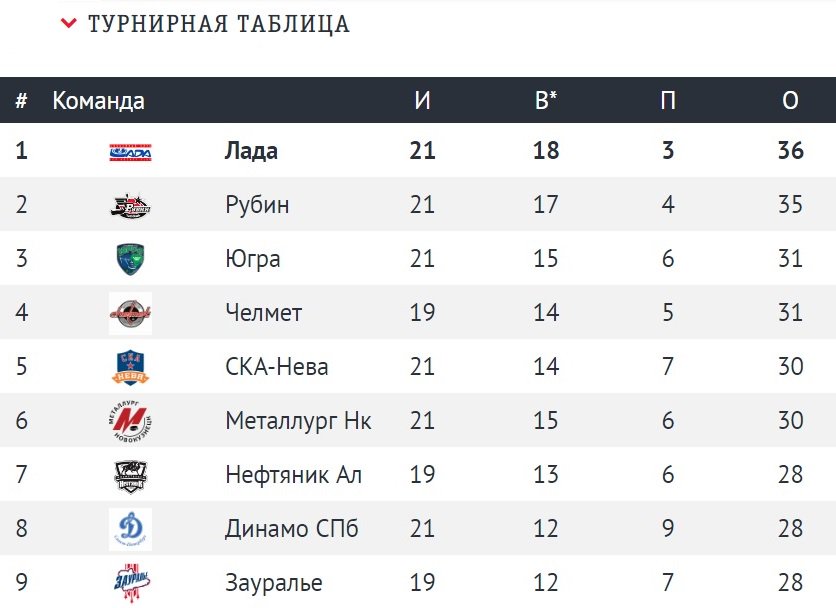
Результаты
Обозначения и предположения
Построенная здесь PN фокусируется на взаимодействиях pVHL, которые уже были подтверждены биохимическими экспериментами и описаны в литературе. Мы решили смоделировать реалистичные пути заболевания VHL на основе подтвержденных литературных данных, включая все известные функции VHL, сигнальный путь, связанный с VHL, и глюкозный метаболизм.Все библиографические источники, использованные для разработки модели, представлены в таблице S1. Окончательный PN состоит из 323 позиций и 238 переходов, соединенных 801 дугой. Таблицы S1 и S2 показывают все места и переходы и связанные с ними биологические соответствия. Места в основном представляют собой белки и ферменты, а некоторые представляют собой ДНК или небольшие молекулярные субстраты, такие как глюкоза и кофакторы (например, АТФ). Обозначения пре- и пост-мест и их биологическое значение объяснены в Таблице S1. В некоторых случаях места используются для представления целой группы изменений, вызванных транскрипцией ДНК (например,г.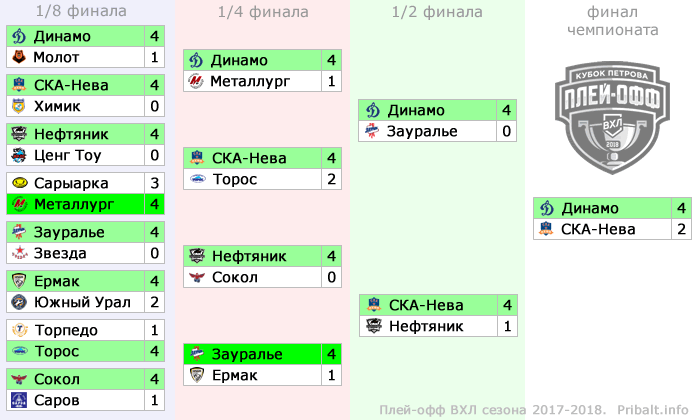 p_32 и p_33 или Et_eff1 и Et_eff2). Вместо этого переходы символизируют комплексообразование между двумя белками или посттрансляционные модификации. Выходные переходы означают деградацию или перемещение в другие части клетки или организма для выполнения своих функций (например, degrad_1 и degrad_2), тогда как входные переходы показывают образование субстрата или белка. Чтобы упростить проектирование такой большой сети, мы решили использовать макроузлы для группировки реакций, представляющих сложные молекулярные пути, такие как сигнальные пути или вторичные сигнальные каскады.Весь процесс объединен в один узел с заданным именем, что позволяет осуществлять визуальный осмотр только в случае необходимости. С верхнего уровня все переходы все еще можно найти на более низком иерархическом уровне макета. Логические узлы использовались для мест, участвующих во многих реакциях по всей сети, таких как ATP и ADP (по 7 логических копий в каждом) или NAD и NADH (по 4 логических копии в каждом). Для моделирования макроузлов была выбрана общая глубина вложенности, равная двум.
p_32 и p_33 или Et_eff1 и Et_eff2). Вместо этого переходы символизируют комплексообразование между двумя белками или посттрансляционные модификации. Выходные переходы означают деградацию или перемещение в другие части клетки или организма для выполнения своих функций (например, degrad_1 и degrad_2), тогда как входные переходы показывают образование субстрата или белка. Чтобы упростить проектирование такой большой сети, мы решили использовать макроузлы для группировки реакций, представляющих сложные молекулярные пути, такие как сигнальные пути или вторичные сигнальные каскады.Весь процесс объединен в один узел с заданным именем, что позволяет осуществлять визуальный осмотр только в случае необходимости. С верхнего уровня все переходы все еще можно найти на более низком иерархическом уровне макета. Логические узлы использовались для мест, участвующих во многих реакциях по всей сети, таких как ATP и ADP (по 7 логических копий в каждом) или NAD и NADH (по 4 логических копии в каждом). Для моделирования макроузлов была выбрана общая глубина вложенности, равная двум. Специальные дуги не использовались, в то время как мы решили смоделировать постоянное присутствие некоторых объектов с помощью двойных дуг (напр.г. для elob, eloc и мест, обозначающих ферментативную активность). В случае белков, которые активно разлагаются, было предпочтительно создать входной переход, имитирующий постоянное производство (или синтез), и выход для потребления. Это относится к pkcz2, Jade1, pVHL и HIF-1α. Как видно из рисунков 1-3, которые представляют всю модель, можно сразу идентифицировать два основных узла: pVHL и vcb, комплекс, образованный pVHL и двумя элонгинами. Другой важной частью является глюкозный метаболизм, смоделированный благодаря его регуляции, индуцированной гипоксией.Подробно он представлен на рисунке 2.
Специальные дуги не использовались, в то время как мы решили смоделировать постоянное присутствие некоторых объектов с помощью двойных дуг (напр.г. для elob, eloc и мест, обозначающих ферментативную активность). В случае белков, которые активно разлагаются, было предпочтительно создать входной переход, имитирующий постоянное производство (или синтез), и выход для потребления. Это относится к pkcz2, Jade1, pVHL и HIF-1α. Как видно из рисунков 1-3, которые представляют всю модель, можно сразу идентифицировать два основных узла: pVHL и vcb, комплекс, образованный pVHL и двумя элонгинами. Другой важной частью является глюкозный метаболизм, смоделированный благодаря его регуляции, индуцированной гипоксией.Подробно он представлен на рисунке 2.
Рисунок 1. Модель верхнего уровня.
Цвета некоторых маркеров были выбраны произвольно, чтобы дать более четкое определение центральных узлов (ATP, Vcb и кислород) или для узлов, участвующих в большем количестве реакций, таких как GSK3β. Группа узлов в левом нижнем углу не отключена от центрального тела сети благодаря наличию логических узлов для синтеза АТФ (t_97).
Группа узлов в левом нижнем углу не отключена от центрального тела сети благодаря наличию логических узлов для синтеза АТФ (t_97).
https://doi.org/10.1371/journal.pone.0096986.g001
Рисунок 3. Нижние иерархические уровни PN, в частности регуляция HIF-1α и HIF-1α-зависимая проангиогенная передача сигналов.
Пути VEGF и EPO находятся на более низком иерархическом уровне, чем макроузел pro_angio.
https://doi.org/10.1371/journal.pone.0096986.g003
Активность транскрипции HIF-1α
Фактор транскрипции HIF-1α стимулирует пролиферацию эндотелиальных клеток для создания новых кровеносных сосудов во время локализованной или обширной гипоксии.У человека он присутствует в виде трех разных паралогов: HIF-1α, HIF-2α и HIF-3α. Последовательность между первыми двумя довольно консервативна, тогда как последняя немного короче и, по-видимому, имеет совершенно другие функции по сравнению с двумя другими [33]–[34]. И HIF-1α, и -2α стимулируют транскрипцию ДНК, но точные продукты этой активности до сих пор плохо изучены. В нашей модели учитывалась только активность HIF-1α in vivo . Не исключено, что от второго паралога зависят и другие биологические эффекты.Действительно, оба имеют проангиогенетическую функцию и расщепляются pVHL посредством пролин-направленного гидроксилирования. HIF представляет собой гетеродимер HIF-1α и HIF-1β, последний также называется ядерным транслокатором рецептора арила (ARNT). Мы начали с транскрипционной активности HIF, поскольку ее регуляция является наиболее изученной функцией pVHL. Наша модель, как и ожидалось на основании литературных данных, показывает, что HIF-1α проникает в ядро, когда он не расщепляется pVHL. Впоследствии он связывает HIF-1β с образованием гетерокомплекса HIF, который взаимодействует с ДНК.Наша модель правильно имитирует повышенное сродство HIF к ДНК. Транскрипция усиливается некоторыми кофакторами, связывающими как субъединицы HIF, так и другие белки, такие как p300, Creb и cjun. Это происходит в специфической последовательности промотора ДНК, называемой элементом реакции на гипоксию (HRE).
В нашей модели учитывалась только активность HIF-1α in vivo . Не исключено, что от второго паралога зависят и другие биологические эффекты.Действительно, оба имеют проангиогенетическую функцию и расщепляются pVHL посредством пролин-направленного гидроксилирования. HIF представляет собой гетеродимер HIF-1α и HIF-1β, последний также называется ядерным транслокатором рецептора арила (ARNT). Мы начали с транскрипционной активности HIF, поскольку ее регуляция является наиболее изученной функцией pVHL. Наша модель, как и ожидалось на основании литературных данных, показывает, что HIF-1α проникает в ядро, когда он не расщепляется pVHL. Впоследствии он связывает HIF-1β с образованием гетерокомплекса HIF, который взаимодействует с ДНК.Наша модель правильно имитирует повышенное сродство HIF к ДНК. Транскрипция усиливается некоторыми кофакторами, связывающими как субъединицы HIF, так и другие белки, такие как p300, Creb и cjun. Это происходит в специфической последовательности промотора ДНК, называемой элементом реакции на гипоксию (HRE). Кроме того, во время транскрипции продуцируются некоторые проангиогенные факторы: фактор роста эндотелия сосудов (VEGF), эндотелин (ET) и эритропоэтин (EPO). Все описанные пути согласуются с предыдущими наблюдениями, опубликованными в [35].
Кроме того, во время транскрипции продуцируются некоторые проангиогенные факторы: фактор роста эндотелия сосудов (VEGF), эндотелин (ET) и эритропоэтин (EPO). Все описанные пути согласуются с предыдущими наблюдениями, опубликованными в [35].
Метаболические процессы
Транскрипционная активность HIF-1α включает некоторые белки, которые зависят от кислорода, но участвуют в других путях (например, в окислительном метаболизме) или полностью независимы (например, металлопротеиназа MT1MMP). Кроме того, HIF-1α стимулирует продукцию белков, участвующих в глюцидном пути. Конечным продуктом метаболизма является аденозинтрифосфат (АТФ), молекулярная форма энергии, состоящая из аденозина, аденинового кольца, соединенного с рибозным сахаром, и трех фосфатных фрагментов.Когда фосфатная часть гидролизуется, она высвобождает энергию, используемую клетками для ферментативных реакций. Метаболизм глюкидов состоит из гликолиза, цикла Кребса, образования гликогена и дыхательной цепи с синтезом АТФ.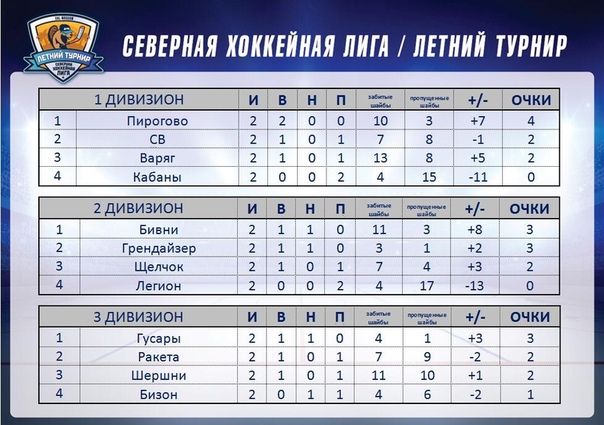 Глюкоза поглощается в клетках ферментативными переносчиками глюкозы (GLUT), которые переносят молекулу в то место внутри клетки, где происходит метаболизм [36]. Существует множество изоформ этих транспортеров: GLUT1 присутствует во всех клетках и, в частности, в мембранах эритроцитов, нейронах и глии [37].GLUT2, расположенный как в печени, так и в бета-клетках поджелудочной железы, характеризуется низким сродством к глюкозе, поэтому для его активации требуется более высокая концентрация глюкозы [38]. Сразу после еды концентрация глюкозы увеличивается, тем самым быстро активируя их. GLUT2 стимулирует выработку инсулина, гормона, регулирующего концентрацию глюкозы в плазме. Концентрация глюкозы в плазме также может увеличиваться за счет противоположного пути, возникающего из-за распада гликогена печени на глюкозу и попадания в системный кровоток.GLUT3 в основном присутствует в нейронах, тогда как GLUT4 является транспортером, активируемым инсулином, расположенным в миоцитах, адипоцитах и кардиомиоцитах [36]–[39].
Глюкоза поглощается в клетках ферментативными переносчиками глюкозы (GLUT), которые переносят молекулу в то место внутри клетки, где происходит метаболизм [36]. Существует множество изоформ этих транспортеров: GLUT1 присутствует во всех клетках и, в частности, в мембранах эритроцитов, нейронах и глии [37].GLUT2, расположенный как в печени, так и в бета-клетках поджелудочной железы, характеризуется низким сродством к глюкозе, поэтому для его активации требуется более высокая концентрация глюкозы [38]. Сразу после еды концентрация глюкозы увеличивается, тем самым быстро активируя их. GLUT2 стимулирует выработку инсулина, гормона, регулирующего концентрацию глюкозы в плазме. Концентрация глюкозы в плазме также может увеличиваться за счет противоположного пути, возникающего из-за распада гликогена печени на глюкозу и попадания в системный кровоток.GLUT3 в основном присутствует в нейронах, тогда как GLUT4 является транспортером, активируемым инсулином, расположенным в миоцитах, адипоцитах и кардиомиоцитах [36]–[39].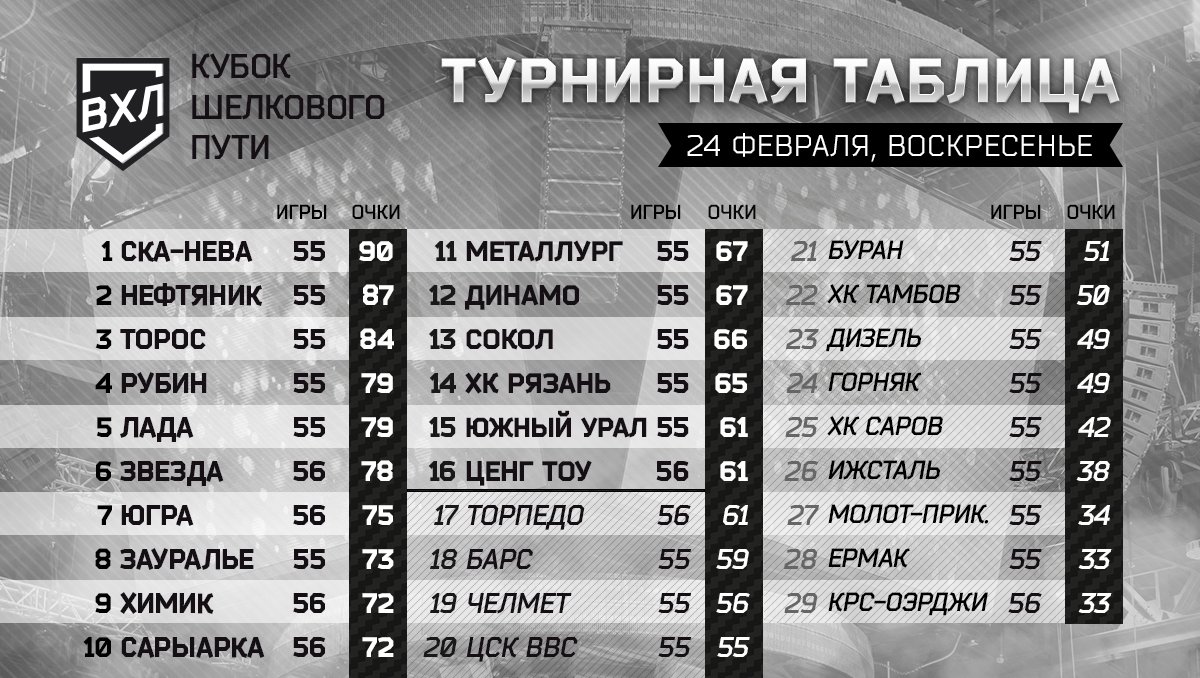 В нашей модели мы решили исключить GLUT3 из-за его специфической роли в нейронных клетках. Гликолиз происходит в цитоплазме, и в ходе этого процесса каждая молекула глюкозы фосфорилируется, потребляя две молекулы АТФ, а затем делится на две более мелкие молекулы. Дальнейшие модификации этих двух молекул приводят к образованию новой АТФ. Молекула, полученная в конце гликолиза, представляет собой пируват, который может быть снова модифицирован тремя различными путями.Он может быть декарбоксилирован и связан с коферментом А с образованием ацетил-кофермента А. Затем его можно карбоксилировать с получением оксалаацетата или трансформировать с помощью лактатдегидрогеназы в молочную кислоту. Пируват также может образовываться другими метаболическими путями, такими как разрушение белков или жирных кислот и модификации аминокислот. Ацетил-КоА и оксалатацетат являются молекулами, используемыми в следующем процессе метаболизма глюкидов, цикле Кребса, происходящем в митохондриальном матриксе. Цикл Кребса начинается со слияния ацетил-КоА и оксалаацетата с образованием лимонной кислоты, которая продолжает подвергаться модификациям до тех пор, пока снова не образуется оксалаксусная кислота.
В нашей модели мы решили исключить GLUT3 из-за его специфической роли в нейронных клетках. Гликолиз происходит в цитоплазме, и в ходе этого процесса каждая молекула глюкозы фосфорилируется, потребляя две молекулы АТФ, а затем делится на две более мелкие молекулы. Дальнейшие модификации этих двух молекул приводят к образованию новой АТФ. Молекула, полученная в конце гликолиза, представляет собой пируват, который может быть снова модифицирован тремя различными путями.Он может быть декарбоксилирован и связан с коферментом А с образованием ацетил-кофермента А. Затем его можно карбоксилировать с получением оксалаацетата или трансформировать с помощью лактатдегидрогеназы в молочную кислоту. Пируват также может образовываться другими метаболическими путями, такими как разрушение белков или жирных кислот и модификации аминокислот. Ацетил-КоА и оксалатацетат являются молекулами, используемыми в следующем процессе метаболизма глюкидов, цикле Кребса, происходящем в митохондриальном матриксе. Цикл Кребса начинается со слияния ацетил-КоА и оксалаацетата с образованием лимонной кислоты, которая продолжает подвергаться модификациям до тех пор, пока снова не образуется оксалаксусная кислота. В процессе модифицируются некоторые коферменты. Декарбоксилирование пирувата с образованием ацетил-КоА уже превращает НАД+ (никотинамидадениндинуклеотид) в НАДН (восстановленную форму), после чего получают еще один из АТФ, ГТФ, ФАДН 2 (флавинадениндинуклеотид) и еще три НАДН на каждую молекулу пирувата, входящую цикл Кребса. Окислительно-восстановительные коферменты считаются переносчиками электронов. В ходе метаболических реакций они восстанавливают себя и получают электроны (и протоны) для окисления субстрата ферментативной реакции.Электроны, захваченные во время метаболизма глюкозы, затем используются в дыхательной цепи, происходящей во внутренней митохондриальной мембране. Дыхательная цепь состоит из транспорта электронов через ферменты, называемые цитохромами, и другие коферменты, характеризующиеся способностью получать и отдавать электроны. НАДН (ФАДН 2 ) снова окисляется цитохромами, возвращаясь в форму НАД (или ФАД). Электроны, полученные в результате окисления, используются для восстановления половины молекулы кислорода до воды, высвобождая больше энергии.
В процессе модифицируются некоторые коферменты. Декарбоксилирование пирувата с образованием ацетил-КоА уже превращает НАД+ (никотинамидадениндинуклеотид) в НАДН (восстановленную форму), после чего получают еще один из АТФ, ГТФ, ФАДН 2 (флавинадениндинуклеотид) и еще три НАДН на каждую молекулу пирувата, входящую цикл Кребса. Окислительно-восстановительные коферменты считаются переносчиками электронов. В ходе метаболических реакций они восстанавливают себя и получают электроны (и протоны) для окисления субстрата ферментативной реакции.Электроны, захваченные во время метаболизма глюкозы, затем используются в дыхательной цепи, происходящей во внутренней митохондриальной мембране. Дыхательная цепь состоит из транспорта электронов через ферменты, называемые цитохромами, и другие коферменты, характеризующиеся способностью получать и отдавать электроны. НАДН (ФАДН 2 ) снова окисляется цитохромами, возвращаясь в форму НАД (или ФАД). Электроны, полученные в результате окисления, используются для восстановления половины молекулы кислорода до воды, высвобождая больше энергии. Окислительно-восстановительная цепь FADH 2 и NADH создает химический потенциал, вызывающий толчок протонов за пределы внутренней мембраны к межмембранному пространству, которое остается между внутренней и внешней мембранами митохондрий. Это также вызывает более высокую концентрацию протонов за пределами внутренней мембраны. Результирующий градиент вызывает стремление протонов войти в клетку. Последним этапом является АТФ-синтетаза, образованная каналом, который позволяет проникать протонам, толкаемым градиентом, позволяя ферменту изменять конформацию и вступать в реакцию.Эта кинетическая энергия превращается в АТФ. В нашей модели подробно описаны гликолитический цикл и цикл Кребса, представленные на втором уровне иерархии грубым переходным гликолизом. Вместо этого дыхательная цепь была объединена в один узел (t_97). Мы решили представить создание и потребление АТФ, чтобы показать влияние более низкой и более высокой концентрации кислорода в сети. С другой стороны, потребление кислорода для синтеза АТФ во время дыхательной цепи создает поток кислорода в модели.
Окислительно-восстановительная цепь FADH 2 и NADH создает химический потенциал, вызывающий толчок протонов за пределы внутренней мембраны к межмембранному пространству, которое остается между внутренней и внешней мембранами митохондрий. Это также вызывает более высокую концентрацию протонов за пределами внутренней мембраны. Результирующий градиент вызывает стремление протонов войти в клетку. Последним этапом является АТФ-синтетаза, образованная каналом, который позволяет проникать протонам, толкаемым градиентом, позволяя ферменту изменять конформацию и вступать в реакцию.Эта кинетическая энергия превращается в АТФ. В нашей модели подробно описаны гликолитический цикл и цикл Кребса, представленные на втором уровне иерархии грубым переходным гликолизом. Вместо этого дыхательная цепь была объединена в один узел (t_97). Мы решили представить создание и потребление АТФ, чтобы показать влияние более низкой и более высокой концентрации кислорода в сети. С другой стороны, потребление кислорода для синтеза АТФ во время дыхательной цепи создает поток кислорода в модели. Кислород — не единственная связь между глюцидным метаболизмом и гипоксией. Действительно, транскрипционная активность HIF-1α усиливает транскрипцию многих изоформ GLUT (таких как 1, 3 и 9) и киназы пируватдегидрогеназы, которая определяет инактивацию пируватдегидрогеназы (PyrDH) и последующее образование ацетил-КоА из пирувата. Наконец, также производится лактатдегидрогеназа, чтобы обеспечить альтернативное соединение, вырабатывающее энергию, необходимую для выживания клеток [36]–[40].
Кислород — не единственная связь между глюцидным метаболизмом и гипоксией. Действительно, транскрипционная активность HIF-1α усиливает транскрипцию многих изоформ GLUT (таких как 1, 3 и 9) и киназы пируватдегидрогеназы, которая определяет инактивацию пируватдегидрогеназы (PyrDH) и последующее образование ацетил-КоА из пирувата. Наконец, также производится лактатдегидрогеназа, чтобы обеспечить альтернативное соединение, вырабатывающее энергию, необходимую для выживания клеток [36]–[40].
pVHL-зависимые процессы
Некоторые интеракторы могут связывать pVHL в областях, взаимодействующих с элонгином C.Это HuR, Nur77, p53 и Jade1. Nur77 имеет сложную функцию, и его роль в pVHL супрессорной активности до сих пор не совсем ясна. Nur77 может связываться с pVHL, ингибируя связывание элонгина, но обеспечивая связывание HIF-1α. Его транскрипция стимулируется самим HIF-1α, а образование комплекса pVHL-HIF-1α-Nur77 стабилизирует транскрипционную активность HIF-1α за счет ингибирования зависимой от pVHL деградации [41]. Другой функцией Nur77 является стимуляция транскрипции проопиомеланокортина (РОМС), который является предшественником образования адренокортикотропного гормона (АКТГ).Этот гормон выполняет важную функцию реакции на стресс, стимулируя выработку кортизола и других нейротрансмиттеров надпочечниками, чтобы усилить реакцию организма на опасность и стрессовые стимулы например. увеличение глюконеогенеза и мышечной массы. Избыток этого гормона может вызвать десенсибилизацию его рецепторов для подавления обратной связи и, следовательно, мышечную слабость, усталость, гипергликемию и остеопороз [42]. p53 может связываться с pVHL, избегая деградации этого супрессора опухоли.Вместо этого он стимулирует апоптотический сигнальный каскад через коактиватор p300, который стимулирует выработку белков, усиливающих запрограммированную гибель клеток. Если p53 не может связываться с pVHL, в модели описаны еще два механизма. Одним из них является его модификация и деградация с помощью Mdm2, а другим является pVHL-независимая деградация HIF-1α.
Другой функцией Nur77 является стимуляция транскрипции проопиомеланокортина (РОМС), который является предшественником образования адренокортикотропного гормона (АКТГ).Этот гормон выполняет важную функцию реакции на стресс, стимулируя выработку кортизола и других нейротрансмиттеров надпочечниками, чтобы усилить реакцию организма на опасность и стрессовые стимулы например. увеличение глюконеогенеза и мышечной массы. Избыток этого гормона может вызвать десенсибилизацию его рецепторов для подавления обратной связи и, следовательно, мышечную слабость, усталость, гипергликемию и остеопороз [42]. p53 может связываться с pVHL, избегая деградации этого супрессора опухоли.Вместо этого он стимулирует апоптотический сигнальный каскад через коактиватор p300, который стимулирует выработку белков, усиливающих запрограммированную гибель клеток. Если p53 не может связываться с pVHL, в модели описаны еще два механизма. Одним из них является его модификация и деградация с помощью Mdm2, а другим является pVHL-независимая деградация HIF-1α. Взаимодействие с Mdm2 необходимо в обоих случаях [43]–[44]. Jade1 является короткоживущим белком, основная функция которого заключается в стимуляции зависимой от фосфорилирования деградации β-катенина.Это субъединица белкового комплекса кадгерина, действующая как внутриклеточный преобразователь сигнала в сигнальном пути Wnt. По-видимому, β-catenin способен останавливать клеточное деление с помощью контактно-зависимого ингибирующего сигнала, тогда как в передаче сигналов Wnt он также участвует в пролиферативной транскрипции. Когда Wnt отсутствует, β-катенин может фосфорилироваться киназой гликогенсинтетазы типа 3β (GSK3β) в комплексе с APC (аденоматозным полипозом кишечной палочки) и аксином. β-catenin может взаимодействовать с Jade1 и успешно разрушаться только после этого взаимодействия [45].Связанные функции представлены в узле макроса Jade1_pat. GSK3β, по-видимому, является белком, участвующим во многих различных путях. GSK3β участвует в дезактивации гликогенсинтетазы и может даже фосфорилировать pVHL и HIF-1α.
Взаимодействие с Mdm2 необходимо в обоих случаях [43]–[44]. Jade1 является короткоживущим белком, основная функция которого заключается в стимуляции зависимой от фосфорилирования деградации β-катенина.Это субъединица белкового комплекса кадгерина, действующая как внутриклеточный преобразователь сигнала в сигнальном пути Wnt. По-видимому, β-catenin способен останавливать клеточное деление с помощью контактно-зависимого ингибирующего сигнала, тогда как в передаче сигналов Wnt он также участвует в пролиферативной транскрипции. Когда Wnt отсутствует, β-катенин может фосфорилироваться киназой гликогенсинтетазы типа 3β (GSK3β) в комплексе с APC (аденоматозным полипозом кишечной палочки) и аксином. β-catenin может взаимодействовать с Jade1 и успешно разрушаться только после этого взаимодействия [45].Связанные функции представлены в узле макроса Jade1_pat. GSK3β, по-видимому, является белком, участвующим во многих различных путях. GSK3β участвует в дезактивации гликогенсинтетазы и может даже фосфорилировать pVHL и HIF-1α..png) В случае HIF-1α он генерирует независимый от pVHL путь деградации, где фосфорилирование делает возможным убиквитинирование, тогда как в случае pVHL он ингибирует стабилизацию pVHL микротрубочек [46].
В случае HIF-1α он генерирует независимый от pVHL путь деградации, где фосфорилирование делает возможным убиквитинирование, тогда как в случае pVHL он ингибирует стабилизацию pVHL микротрубочек [46].
Анализ структурной модели
Также на основе предыдущих наблюдений Heiner et al., [47], в 2008 г. Grunwald et al. продемонстрировали, что PN можно использовать для описания больших и сложных метаболических путей [31]. Они постулировали следующий набор минимальных правил, которым должна удовлетворять СП, чтобы считаться биологически надежной: (1) сеть должна быть полностью связанной, (2) сеть должна быть покрыта Т-инвариантами и (3) каждый Т-инвариант и P-инвариант должен иметь биологический смысл. Описанная здесь модель была протестирована относительно того, что ранее было сделано Грюнвальдом и его сотрудниками [31], и в результате она покрылась Т-инвариантами, связна, однородна, и каждое место имеет предпереход и постпереход.Переходы без пре- или пост-мест использовались для имитации интерфейса системы с окружением. Сеть живая, другими словами, она продолжает работать вечно, при этом все переходы навсегда вносят вклад в поведение сети, а мертвых переходов нет. MCTS и кластерный анализ были использованы из-за большого количества Т- и Р-инвариантов, включенных в модель. Оба метода используются в теории СП, чтобы уменьшить сложность, связанную с такой большой сетью, и уменьшить количество ошибок, связанных с ручным исследованием.Из 238 переходов, присутствующих в начале в модели, было вычислено 393 Т-инварианта без учета 10 тривиальных инвариантов. Последние состоят из пары переходов, которые обычно представляют собой прямую и обратную реакцию, например активное и неактивное состояние белка. Тривиальные инварианты могут быть стерты, чтобы уменьшить размер сети, не нарушая всей системы, когда интерес сосредоточен на стационарном поведении [12]. Т-инварианты были сгруппированы в 44 кластера с использованием коэффициента Танимото с порогом подобия 65%, как описано в [31].Только 11 из этих 44 содержали более одного Т-инварианта.
Сеть живая, другими словами, она продолжает работать вечно, при этом все переходы навсегда вносят вклад в поведение сети, а мертвых переходов нет. MCTS и кластерный анализ были использованы из-за большого количества Т- и Р-инвариантов, включенных в модель. Оба метода используются в теории СП, чтобы уменьшить сложность, связанную с такой большой сетью, и уменьшить количество ошибок, связанных с ручным исследованием.Из 238 переходов, присутствующих в начале в модели, было вычислено 393 Т-инварианта без учета 10 тривиальных инвариантов. Последние состоят из пары переходов, которые обычно представляют собой прямую и обратную реакцию, например активное и неактивное состояние белка. Тривиальные инварианты могут быть стерты, чтобы уменьшить размер сети, не нарушая всей системы, когда интерес сосредоточен на стационарном поведении [12]. Т-инварианты были сгруппированы в 44 кластера с использованием коэффициента Танимото с порогом подобия 65%, как описано в [31].Только 11 из этих 44 содержали более одного Т-инварианта. Три самых больших кластера — это C9, состоящий из 144 T-инвариантов, C8 из 72 и C11 из 64 T-инвариантов. Разделение на кластеры позволяет упростить анализ сетевых путей, представленных каждым Т-инвариантом, поскольку они сгруппированы по сходству, в частности, по общим переходам, из которых они состоят. Т-инварианты, названные в тексте, показаны в таблице S3, а Т-инварианты, сгруппированные в C8, C9, C10, C11, объяснены в таблице S4 и описаны следующим образом.
Три самых больших кластера — это C9, состоящий из 144 T-инвариантов, C8 из 72 и C11 из 64 T-инвариантов. Разделение на кластеры позволяет упростить анализ сетевых путей, представленных каждым Т-инвариантом, поскольку они сгруппированы по сходству, в частности, по общим переходам, из которых они состоят. Т-инварианты, названные в тексте, показаны в таблице S3, а Т-инварианты, сгруппированные в C8, C9, C10, C11, объяснены в таблице S4 и описаны следующим образом.
Кластер C8
Кластер C8 объединяет все переходы, включенные в пути HIF-1α, включая транскрипцию, сигнальные каскады, деградацию через pVHL, p53 и GSK3β и, в конечном итоге, цикл Кребса. Для сигнального пути ЭПО не включены два перехода (t_35 и t_36), которые вызывают активацию Jak и последующую активацию Stat5 для стимуляции транскрипции ДНК. Регуляция стабильности матрикса также является частью кластера из-за дестабилизации, вызванной HIF-1α транскрипцией металлопротеиназы (MMP), переходами от t_134 к t_140. Самый большой T-инвариант в C8 — это Inv_280 (93 перехода), а наименьший — Inv_377 (81 переход). Различия между Т-инвариантами показывают возможность альтернативных путей внутри модели. Например, сигнальный каскад, зависимый от VEGF, может протекать тремя различными способами: t_13, t_14 и t_15, что приводит к слиянию путей в грубых узлах Vegf_path4, Vegf_path3 и Vegf_path2 соответственно. Частота возникновения в C8 составляет 24 перехода для каждого пути. Пути эндотелина, VEGF и эритропоэтина не конфликтуют и происходят вместе.Дезагрегация матрикса посредством ММР присутствует в 18 Т-инвариантах, тогда как ингибирование этих белков, т.е. стабилизация матрикса, присутствует в остальных 54 переходах. Что касается цикла Кребса, 47 Т-инвариантов имеют t_91, из которых только 24 достигают t_92 и t_93, представляющих последние три стадии цикла: от сукцината до фумарата, от фумарата до малата и от малата до оксалоацетата. Весь производимый малат используется для регенерации оксалоацетата. Деградация HIF-1α происходит в любом Т-инварианте кластера.
Самый большой T-инвариант в C8 — это Inv_280 (93 перехода), а наименьший — Inv_377 (81 переход). Различия между Т-инвариантами показывают возможность альтернативных путей внутри модели. Например, сигнальный каскад, зависимый от VEGF, может протекать тремя различными способами: t_13, t_14 и t_15, что приводит к слиянию путей в грубых узлах Vegf_path4, Vegf_path3 и Vegf_path2 соответственно. Частота возникновения в C8 составляет 24 перехода для каждого пути. Пути эндотелина, VEGF и эритропоэтина не конфликтуют и происходят вместе.Дезагрегация матрикса посредством ММР присутствует в 18 Т-инвариантах, тогда как ингибирование этих белков, т.е. стабилизация матрикса, присутствует в остальных 54 переходах. Что касается цикла Кребса, 47 Т-инвариантов имеют t_91, из которых только 24 достигают t_92 и t_93, представляющих последние три стадии цикла: от сукцината до фумарата, от фумарата до малата и от малата до оксалоацетата. Весь производимый малат используется для регенерации оксалоацетата. Деградация HIF-1α происходит в любом Т-инварианте кластера. Зависимая от pVHL деградация HIF-1α всегда присутствует (переходы с t_116 на t_119). В 19 Т-инвариантах деградация происходит через р53 (от t_191 до t_193) или, альтернативно, посредством фосфорилирования с помощью GSK3β в других 17 Т-инвариантах. Два из трех путей могут присутствовать в одном и том же Т-инварианте, как и в Inv_227, где присутствуют как деградация через pVHL, так и деградация через p53. Это рассматривалось как зависимость HIF-1α от отсутствия деградации этими белками. Все три пути деградации никогда не проявляются в одном и том же Т-инварианте.Пути p53 и GSK3β никогда не присутствуют вместе, но каждый из них сопровождается зависимой от pVHL протеасомной деградацией. У Inv_377 отсутствует сигнальный путь ЭПО, но он единственный в этом кластере имеет t_34, t_33 и t_37. Эти инварианты имеют все входные и выходные переходы. Например, t_202, второй вход для pVHL, присутствует только в 18 инвариантах. Другими входами являются t_98, всегда присутствующий, приводящий к образованию HIF-1α и pVHL, t_192, продуцирующий p53 и t_216, представляющий другие метаболические пути, генерирующие пируват.
Зависимая от pVHL деградация HIF-1α всегда присутствует (переходы с t_116 на t_119). В 19 Т-инвариантах деградация происходит через р53 (от t_191 до t_193) или, альтернативно, посредством фосфорилирования с помощью GSK3β в других 17 Т-инвариантах. Два из трех путей могут присутствовать в одном и том же Т-инварианте, как и в Inv_227, где присутствуют как деградация через pVHL, так и деградация через p53. Это рассматривалось как зависимость HIF-1α от отсутствия деградации этими белками. Все три пути деградации никогда не проявляются в одном и том же Т-инварианте.Пути p53 и GSK3β никогда не присутствуют вместе, но каждый из них сопровождается зависимой от pVHL протеасомной деградацией. У Inv_377 отсутствует сигнальный путь ЭПО, но он единственный в этом кластере имеет t_34, t_33 и t_37. Эти инварианты имеют все входные и выходные переходы. Например, t_202, второй вход для pVHL, присутствует только в 18 инвариантах. Другими входами являются t_98, всегда присутствующий, приводящий к образованию HIF-1α и pVHL, t_192, продуцирующий p53 и t_216, представляющий другие метаболические пути, генерирующие пируват. Последний также присутствует в каждом инварианте, обеспечивающем образование пирувата, необходимого для прохождения цикла Кребса.
Последний также присутствует в каждом инварианте, обеспечивающем образование пирувата, необходимого для прохождения цикла Кребса.
Кластер C9
КластерC9 является самым большим кластером в нашей модели и включает 144 Т-инварианта. Для него характерна полная отмена пути ЭПО, который проходит через образование комплекса Shc-Grb-Sos и последующий мапк-зависимый каскад фосфорилирования. Переход t_127, отражающий влияние ЭПО на выработку кислорода, отсутствует. Вместо него рассматриваются t_35 и t_36, которые присутствуют в 72 Т-инвариантах.В кластере C9 самыми большими Т-инвариантами являются Inv_278 и Inv_279 (74 перехода), а самыми короткими являются Inv_101, Inv_105, Inv_144 и Inv_148 с 65 переходами в каждом.
Кластер C10
Кластер С10, состоящий из 52 Т-инвариантов, характеризуется наличием гликолиза между многими переходами, сгруппированными в кластере. Это также кластер, содержащий наиболее заполненный Т-инвариант из всех вычисленных 393 нетривиальных Т-инвариантов. Это Inv_245, включающий 101 переход и покрывающий почти половину всей модели.Кластер C10 также включает Inv_125, самый короткий инвариант этой модели, состоящий из 85 переходов из-за отсутствия цикла Кребса. Еще одно отличие от трех других основных кластеров состоит в том, что здесь присутствуют оба пути ЭПО, в частности, путь Jak относится к 4 Т-инвариантам, а Shc-Grb-Sos наблюдается во всем кластере. Vegf_path2, по-видимому, более распространен в этом кластере, поскольку присутствует в 36 T-инвариантах, тогда как два других присутствуют по 12 раз каждый. На этот раз они присутствуют даже в том же инварианте, что и для Inv_60, Inv_129, Inv_172 и Inv_215, причем как t_13 и t_15, так и Inv_142, Inv_185 и Inv_228 с t_14 и t_15 и всеми последующими сигналами, появляющимися одновременно.Несмотря на то, что гликолиз присутствует во всех кластерных инвариантах, цикл Кребса проявляется только в 11 случаях. p53-зависимая деградация HIF-1α происходит в 11 случаях, а зависимая от фосфорилирования — в 13.
Это Inv_245, включающий 101 переход и покрывающий почти половину всей модели.Кластер C10 также включает Inv_125, самый короткий инвариант этой модели, состоящий из 85 переходов из-за отсутствия цикла Кребса. Еще одно отличие от трех других основных кластеров состоит в том, что здесь присутствуют оба пути ЭПО, в частности, путь Jak относится к 4 Т-инвариантам, а Shc-Grb-Sos наблюдается во всем кластере. Vegf_path2, по-видимому, более распространен в этом кластере, поскольку присутствует в 36 T-инвариантах, тогда как два других присутствуют по 12 раз каждый. На этот раз они присутствуют даже в том же инварианте, что и для Inv_60, Inv_129, Inv_172 и Inv_215, причем как t_13 и t_15, так и Inv_142, Inv_185 и Inv_228 с t_14 и t_15 и всеми последующими сигналами, появляющимися одновременно.Несмотря на то, что гликолиз присутствует во всех кластерных инвариантах, цикл Кребса проявляется только в 11 случаях. p53-зависимая деградация HIF-1α происходит в 11 случаях, а зависимая от фосфорилирования — в 13. Был добавлен входной переход по отношению к другим основным кластерам, проанализированным до сих пор (например, t_69_eating), без которого гликолиз никогда не мог бы иметь место.
Был добавлен входной переход по отношению к другим основным кластерам, проанализированным до сих пор (например, t_69_eating), без которого гликолиз никогда не мог бы иметь место.
Кластер C11
Кластер C11 состоит из 64 Т-инвариантов. Здесь описана только часть пути ЭПО, с тем основным отличием, что цикл Кребса полностью отменен, в то время как включена регуляция пролилгидроксилазы типа 2 (PHD2) с помощью оксалацетата.Взаимодействие HIF-1α с Nur77 и транскрипция VEGF с помощью Sp1 также присутствуют. t_80 (превращение пирувата в оксалатацетат) отсутствует в первых 42 кластерных Т-инвариантах. Взаимодействие Nur77 с HIF-1α присутствует только в 8 Т-инвариантах, в частности, от Inv_87 до Inv_94. Транскрипционная активность Sp1 проявляется в двойном количестве, включая те же 8 только что упомянутых инвариантов. Транскрипция VEGF через активность Sp1 зависит от фосфорилирования aPKCζ2, которое, однако, не проявляется в кластере.Когда VEGF синтезируется, он впоследствии стабилизируется с помощью Hur, за которым следует t_178, и Hur воссоздается, чтобы обеспечить другие функции. Действительно, это одно из немногих мест без перехода ввода, но с токеном, который снова идет вперед и назад. По сравнению с другими кластерами, C11 также показывает на один переход меньше в крупном узле регуляции PHD, в частности, t_81, который показывает превращение пирувата под действием pyrDH в ацетил-Кофермент А, необходимый для цикла Кребса. Четыре кластера от C-8 до C12 очень похожи друг на друга, как видно из дерева расстояний на рисунке 4.Все они содержат транскрипционную активность HIF-1α и сигнальные пути, вызванные вариантами деградации EPO, VEGF и HIF-1α. Они включают эффекты других продуктов транскрипционной активности, таких как металлопротеиназа и киназа пируватдегидрогеназы, которые регулируют состояние активации PyrDH. Все они включают часть глюкозного метаболизма, но не само образование гликогена. Остальные пять кластеров от С12 до С16 имеют меньшее количество Т-инвариантов и меньшее количество переходов в каждом инварианте.Они не включают транскрипционную активность, а образуются только VEGF и гликолитическими путями.
Действительно, это одно из немногих мест без перехода ввода, но с токеном, который снова идет вперед и назад. По сравнению с другими кластерами, C11 также показывает на один переход меньше в крупном узле регуляции PHD, в частности, t_81, который показывает превращение пирувата под действием pyrDH в ацетил-Кофермент А, необходимый для цикла Кребса. Четыре кластера от C-8 до C12 очень похожи друг на друга, как видно из дерева расстояний на рисунке 4.Все они содержат транскрипционную активность HIF-1α и сигнальные пути, вызванные вариантами деградации EPO, VEGF и HIF-1α. Они включают эффекты других продуктов транскрипционной активности, таких как металлопротеиназа и киназа пируватдегидрогеназы, которые регулируют состояние активации PyrDH. Все они включают часть глюкозного метаболизма, но не само образование гликогена. Остальные пять кластеров от С12 до С16 имеют меньшее количество Т-инвариантов и меньшее количество переходов в каждом инварианте.Они не включают транскрипционную активность, а образуются только VEGF и гликолитическими путями.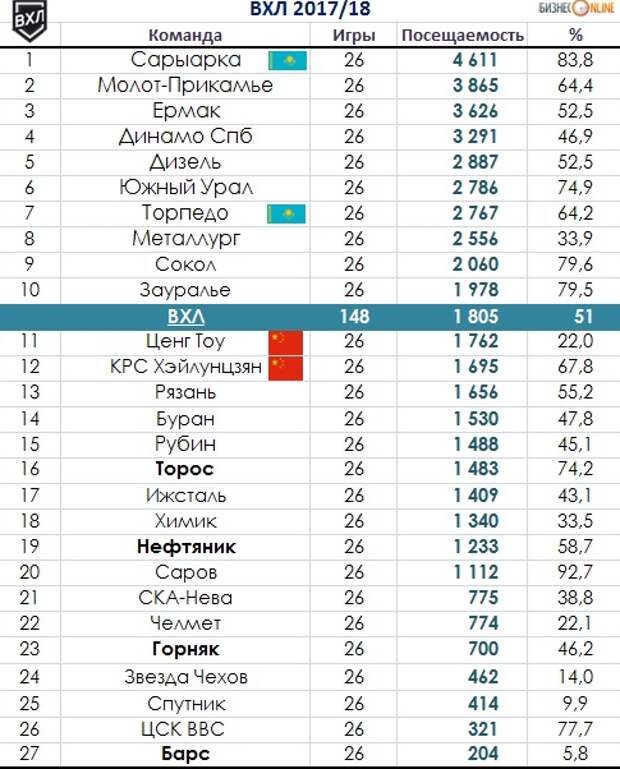 Информативность этих кластеров оказалась малоинформативной и их анализ не проводился. То же самое относится и к кластерам, состоящим из 1–3 Т-инвариантов. Наконец, некоторые переходы отсутствуют в кластерах и не перечислены в Т-инвариантах, поскольку тривиальные инварианты были исключены из кластерного анализа. Эти переходы показаны в таблице 1 с их соответствующим биологическим значением.
Информативность этих кластеров оказалась малоинформативной и их анализ не проводился. То же самое относится и к кластерам, состоящим из 1–3 Т-инвариантов. Наконец, некоторые переходы отсутствуют в кластерах и не перечислены в Т-инвариантах, поскольку тривиальные инварианты были исключены из кластерного анализа. Эти переходы показаны в таблице 1 с их соответствующим биологическим значением.
Анализ MCTS
Другой способ группировки инвариантов — по количеству присутствующих в них одиночных переходов. Анализ максимального общего набора переходов (MCTS) обеспечивает декомпозицию PN на непересекающиеся подсети, разделяющие части одних и тех же Т-инвариантов [29]. В биохимической сети MCTS можно интерпретировать как подмножества ферментов, работающих вместе в устойчивых условиях, рассчитанных на основе поддержки Т-инварианта. Расчет MCTS не учитывает стехиометрические отношения, описывая исключительно наборы реакций, представленных максимальным числом Т-инвариантов, которые являются общими для разных сигнальных путей [48].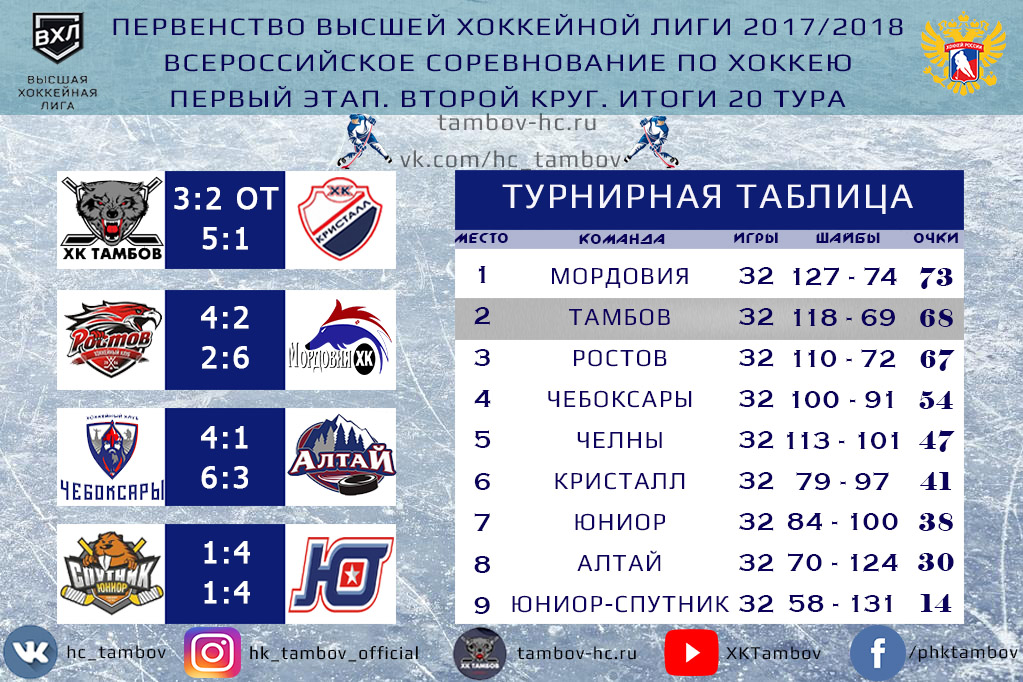 Всего было идентифицировано 40 нетривиальных MCTS, результаты и связанные с ними биологические средства показаны в таблице 2. Некоторые переходы не относятся к нетривиальным MCTS, поскольку их возникновение не имеет сходства с другими переходами и они создают отдельные MCTS (в частности, : t_69, t_82, t_91, t_94, t_98, t_114, t_116, t_120, t_121, t_122, t_179, t_202, t_209, t_212, t_216, t_225 и t_229). MCTS определяют переходы, которые всегда происходят вместе, но не обязательно связаны между собой, таким образом, представляя разрозненные строительные блоки, составляющие сеть.С учетом обоих анализов в PInA [29] была автоматически построена таблица, показывающая корреляцию между кластерами и MCTS. Переходы (t) или MCTS (M) сравнивают, чтобы оценить, сколько кластеров T-инвариантов покрывают выбранные M или t (если переход еще не является частью MCTS, как указано выше). Чем более охвачен переход или набор, тем более важным он может считаться для поведения сети. Недавно был представлен метод укрупнения сети, основанный на абстрактных зависимых наборах переходов (ADT) [49].
Всего было идентифицировано 40 нетривиальных MCTS, результаты и связанные с ними биологические средства показаны в таблице 2. Некоторые переходы не относятся к нетривиальным MCTS, поскольку их возникновение не имеет сходства с другими переходами и они создают отдельные MCTS (в частности, : t_69, t_82, t_91, t_94, t_98, t_114, t_116, t_120, t_121, t_122, t_179, t_202, t_209, t_212, t_216, t_225 и t_229). MCTS определяют переходы, которые всегда происходят вместе, но не обязательно связаны между собой, таким образом, представляя разрозненные строительные блоки, составляющие сеть.С учетом обоих анализов в PInA [29] была автоматически построена таблица, показывающая корреляцию между кластерами и MCTS. Переходы (t) или MCTS (M) сравнивают, чтобы оценить, сколько кластеров T-инвариантов покрывают выбранные M или t (если переход еще не является частью MCTS, как указано выше). Чем более охвачен переход или набор, тем более важным он может считаться для поведения сети. Недавно был представлен метод укрупнения сети, основанный на абстрактных зависимых наборах переходов (ADT) [49]. Он сформулирован без требования предварительного вычисления Т-инвариантов и является инструментом, обычно используемым для разложения больших биохимических сетей на более мелкие подсети. Из-за ручной разработки нашей модели мы предпочли поддерживать логическую иерархию, основанную на метаболических путях, чтобы поддерживать сеть, сосредоточенную на pVHL и ее взаимодействии. Результаты расчета MCTS показывают, что набор, наиболее покрытый T-инвариантами кластера, — это M20 с 358 T-инвариантами, покрывающими все переходы в наборе, что указывает на то, что этот MCTS соответствует большему количеству T-инвариантов, чем другие.Все наборы переходов являются важным связующим звеном с остальными, так как токены чаще проходят через эти переходы. Переход, не присутствующий ни в одном наборе, но наиболее покрываемый Т-инвариантами, — это t_98, который также является наиболее часто встречающимся переходом, см. рис. 5. 10 наиболее часто встречающихся переходов перечислены в таблице 3.
Он сформулирован без требования предварительного вычисления Т-инвариантов и является инструментом, обычно используемым для разложения больших биохимических сетей на более мелкие подсети. Из-за ручной разработки нашей модели мы предпочли поддерживать логическую иерархию, основанную на метаболических путях, чтобы поддерживать сеть, сосредоточенную на pVHL и ее взаимодействии. Результаты расчета MCTS показывают, что набор, наиболее покрытый T-инвариантами кластера, — это M20 с 358 T-инвариантами, покрывающими все переходы в наборе, что указывает на то, что этот MCTS соответствует большему количеству T-инвариантов, чем другие.Все наборы переходов являются важным связующим звеном с остальными, так как токены чаще проходят через эти переходы. Переход, не присутствующий ни в одном наборе, но наиболее покрываемый Т-инвариантами, — это t_98, который также является наиболее часто встречающимся переходом, см. рис. 5. 10 наиболее часто встречающихся переходов перечислены в таблице 3.
P-инвариантный анализ
Хотя сеть не покрыта P-инвариантами, она имеет 130 P-инвариантов. 47 из них являются тривиальными P-инвариантами, состоящими из одного места, соединенного двойными дугами, чтобы имитировать функцию дуги активатора.Другой объект, представленный двойными дугами, представляет собой ферментативную активность, катализирующую реакцию и немедленно возвращающуюся к устойчивому состоянию. P-инварианты показывают места или наборы мест, где номера токенов всегда остаются одинаковыми и не выходят за пределы подсети, индуцированной P-инвариантом в исходной маркировке. Другими словами, они не растут и не уменьшаются. Остальные P-инварианты в основном расположены в путях передачи сигнала, например, в ситуациях, когда белок отключается от своей функции, а затем возвращается после второго механизма реактивации.Этот сценарий присутствует в p_41, p_42 и p_45, расположенных в инварианте P_58. Важно отметить, что АТФ и АДФ, а также НАД и НАДН моделируются как Р-инварианты. P_90, P_91 и t_97 способны трансформировать АТФ и АДФ. В более общем случае все переходы с потреблением энергии считаются обратными переходами инвариантов.
47 из них являются тривиальными P-инвариантами, состоящими из одного места, соединенного двойными дугами, чтобы имитировать функцию дуги активатора.Другой объект, представленный двойными дугами, представляет собой ферментативную активность, катализирующую реакцию и немедленно возвращающуюся к устойчивому состоянию. P-инварианты показывают места или наборы мест, где номера токенов всегда остаются одинаковыми и не выходят за пределы подсети, индуцированной P-инвариантом в исходной маркировке. Другими словами, они не растут и не уменьшаются. Остальные P-инварианты в основном расположены в путях передачи сигнала, например, в ситуациях, когда белок отключается от своей функции, а затем возвращается после второго механизма реактивации.Этот сценарий присутствует в p_41, p_42 и p_45, расположенных в инварианте P_58. Важно отметить, что АТФ и АДФ, а также НАД и НАДН моделируются как Р-инварианты. P_90, P_91 и t_97 способны трансформировать АТФ и АДФ. В более общем случае все переходы с потреблением энергии считаются обратными переходами инвариантов. Инварианты, не связанные с передачей сигнала, представляют собой места, расположенные в системе Hur, где Hur отстранен от своей функции с помощью pVHL. Это хорошее приближение для последовательных модификаций, которые мгновенно активируют белки.После этого Hur может вернуться и стабилизировать VEGF, чтобы повысить его транскрипционную активность.
Инварианты, не связанные с передачей сигнала, представляют собой места, расположенные в системе Hur, где Hur отстранен от своей функции с помощью pVHL. Это хорошее приближение для последовательных модификаций, которые мгновенно активируют белки.После этого Hur может вернуться и стабилизировать VEGF, чтобы повысить его транскрипционную активность.
Эксперименты с нокаутом In Silico
Описанные ранее кластеризация и анализ MCTS для Т-инвариантов позволили нам выявить наиболее распространенные переходы и понять, какие переходы можно истощать в наших экспериментах с нокаутом, чтобы получить наиболее важный биологический эффект. Были проведены эксперименты по отключению, стирая выбранные переходы или токены и наблюдая, какие переходы или MCTS становятся инактивированными.Принимая во внимание наши результаты и данные литературы, мы решили исключить следующие элементы пути: (i) pVHL, (ii) только HIF1α и с Sp1, (iii) t_98, (iv) PHD2, (v) MCTS1, (vi) t_97 и (vii) GSK3β. Далее мы опишем влияние каждого сценария нокаута на нашу модель.
(i) нокаут pVHL.
Деградация HIF-1α не полностью истощена из-за наличия как p53-, так и GSK3β-зависимых альтернативных путей деградации. Все другие процессы, обычно ингибируемые pVHL, происходят неконтролируемым образом, включая создание VEGF за счет транскрипционной активности Sp1 и усиление регуляции матрикса из-за отсутствия поперечного связывания фибронектина.В результате Hur постоянно активируется, а nur77 может стимулировать синтез проопиомеланокортина, предшественника адренокортикотропного гормона. Card9 увеличивает высвобождение фактора некроза опухоли и NF-kB, если он не ингибируется pVHL. Вместо этого Jade1 не может выживать достаточно долго, чтобы ингибировать β-катенин, генерируя сигнал пролиферации с помощью Wnt. Молочная кислота также не вырабатывается из-за того, что продукция фермента ЛДГ зависит от транскрипционной активности HIF-1α.
(ii) нокаут HIF-1α.
VEGF все еще вырабатывается благодаря Sp1, поэтому кислород все еще вырабатывается, хотя и в меньшей пропорции. Если HIF-1α и Sp1 нокаутированы одновременно, кислород быстро потребляется, и метаболизм вскоре не может продолжаться. Молочная кислота не вырабатывается из-за того, что продукция фермента ЛДГ зависит от транскрипционной активности HIF-1α. Гликолиз и гликоген вырабатываются нормально, и метаболизм не ингибируется отрицательной регуляцией PyrDH и образованием молочной кислоты. Поскольку pVHL присутствует, активируются другие супрессорные активности, за исключением протеасомной деградации HIF-1α из-за отсутствия субстрата.
Если HIF-1α и Sp1 нокаутированы одновременно, кислород быстро потребляется, и метаболизм вскоре не может продолжаться. Молочная кислота не вырабатывается из-за того, что продукция фермента ЛДГ зависит от транскрипционной активности HIF-1α. Гликолиз и гликоген вырабатываются нормально, и метаболизм не ингибируется отрицательной регуляцией PyrDH и образованием молочной кислоты. Поскольку pVHL присутствует, активируются другие супрессорные активности, за исключением протеасомной деградации HIF-1α из-за отсутствия субстрата.
(iii) Двойной нокаут HIF-1α и pVHL.
Это создает ситуацию, когда метаболизм нормальный, но регенерация кислорода менее продуктивна, и только Sp1 действует на транскрипцию. Из-за отсутствия pVHL все процессы, стимулирующие пролиферацию, активны, что приводит к несбалансированному потреблению ресурсов. Наша модель показывает, что это состояние совместимо с ростом и размножением клеток, но образование новых кровеносных сосудов происходит значительно медленнее, а глюкозный метаболизм в основном основан на реакции гликолиза. Аналогичное снижение активности относится как к регуляции пути плотных контактов, так и к клеточному внешнему матриксу (ECM). Нельзя исключить, что некоторые наблюдаемые эффекты могут быть смягчены активностью как HIF-2α, так и HIF-3α in vivo.
Аналогичное снижение активности относится как к регуляции пути плотных контактов, так и к клеточному внешнему матриксу (ECM). Нельзя исключить, что некоторые наблюдаемые эффекты могут быть смягчены активностью как HIF-2α, так и HIF-3α in vivo.
(iv) Выбивной PHD2.
Белок участвует в опосредованной pVHL и зависимой от кислорода деградации HIF-1α. Кроме того, PHD2 участвует в гидроксилировании субъединицы Rpb1 РНК-полимеразы II, что делает возможным ее транслокацию в области ядра с меньшей концентрацией хроматина.Когда он нокаутирован, деградация HIF-1α может продолжаться альтернативными путями, как показано в эксперименте с нокаутом pVHL, и активность РНК-полимеразы II выше, даже если rpb7 все еще может быть инактивирован pVHL.
(v) Выбивание MCTS1.
MCTS1 объединяет некоторые реакции, участвующие в пути р53-зависимой деградации HIF-1α (таблица 2). Чтобы выполнить это отключение, мы стерли необходимый маркер в mdm2, сделав предварительное условие недостаточным для включения переходов MCTS. p53 не деградирует и может продолжать свой проапоптотический сигнал.С другой стороны, механизм деградации HIF-1α также отключается, что приводит к повышенной транскрипционной активности HIF-1α.
p53 не деградирует и может продолжать свой проапоптотический сигнал.С другой стороны, механизм деградации HIF-1α также отключается, что приводит к повышенной транскрипционной активности HIF-1α.
(vi) t_97 нокаут.
Это переход АТФазы, позволяющий модели имитировать потребление кислорода для синтеза АТФ. Если этот переход неактивен, кислород накапливается бесконечно, и АТФ не регенерируется после нескольких шагов моделирования. Вначале АТФ образуется на первом этапе гликолиза, но затем снова расходуется. В какой-то момент в этих реакциях нет доступного АТФ, чтобы позволить системе восстановить баланс потребляемого АТФ.После нескольких шагов моделирования кислород достигает высокого уровня из-за более медленного потребления в процессе регулирования PHD2. Биологически это означает, что обмен веществ останавливается, и клетка не способна создавать энергию для выживания. В модели нет накопления, кроме глюкозы. Некоторые процессы создания кислорода также заблокированы из-за отсутствия АТФ, например. т_15, т_41 и т_57.
т_15, т_41 и т_57.
(vii) Выбивание GSK3β.
Этот фермент участвует в отрицательной регуляции гликогенсинтетазы (GS) и инактивируется при фосфорилировании.Когда GSK3β нокаутирован, гликоген непрерывно вырабатывается за счет того, что фермент остается в активном состоянии. В реальном организме существуют альтернативные формы GSK3β, способные инактивировать ГС, поэтому эффект будет менее резким. GSK3β также участвует в деградации HIF-1α, вызывая его фосфорилирование и последующее убиквитинирование. Он также участвует в деградации β-катенина, где отвечает за первичное фосфорилирование. В случае нокаута, даже если Jade1 может быть стабилизирован с помощью pVHL, эффект будет аналогичен нокауту Jade1, где β-catenin свободен для продолжения пролиферации, стимулируя транскрипционную активность.
Обсуждение
Мы начали с базовой модели реакции на гипоксию [12] и расширили исходную сеть функциональными данными, полученными из литературы, чтобы представить полное описание пути взаимодействия pVHL в соответствии с текущими знаниями. Синдром ВХЛ характеризуется образованием опухолей и кист, поражающих различные отделы и ткани организма. Действительно, pVHL является опухолевым супрессором, функции которого связаны с ингибированием пролиферации и выживания, роста и стабильности внеклеточного матрикса и микротрубочек, а также клеточной полярности и миграции.База данных IntAct сообщает о более чем 200 предполагаемых взаимодействующих с pVHL, и для большинства из них детали взаимодействия и функции остаются в значительной степени неизвестными. Мы решили смоделировать взаимодействия pVHL в достоверном клеточном контексте, при этом активность многих белков происходит одновременно. Основная идея состояла в том, чтобы создать новое вручную составленное описание PN всего пути заболевания VHL, включая глюкозный метаболизм и сигнальные пути. Модель спроектирована как стандартная PN и состоит из 238 переходов и 323 мест, соединенных 801 ребром.Биологически реалистичная модель PN должна быть покрыта T-инвариантами, что означает, что каждый переход в модели должен быть включен в T-инвариант, и каждый инвариант должен иметь биологическое значение [31]–[47].
Синдром ВХЛ характеризуется образованием опухолей и кист, поражающих различные отделы и ткани организма. Действительно, pVHL является опухолевым супрессором, функции которого связаны с ингибированием пролиферации и выживания, роста и стабильности внеклеточного матрикса и микротрубочек, а также клеточной полярности и миграции.База данных IntAct сообщает о более чем 200 предполагаемых взаимодействующих с pVHL, и для большинства из них детали взаимодействия и функции остаются в значительной степени неизвестными. Мы решили смоделировать взаимодействия pVHL в достоверном клеточном контексте, при этом активность многих белков происходит одновременно. Основная идея состояла в том, чтобы создать новое вручную составленное описание PN всего пути заболевания VHL, включая глюкозный метаболизм и сигнальные пути. Модель спроектирована как стандартная PN и состоит из 238 переходов и 323 мест, соединенных 801 ребром.Биологически реалистичная модель PN должна быть покрыта T-инвариантами, что означает, что каждый переход в модели должен быть включен в T-инвариант, и каждый инвариант должен иметь биологическое значение [31]–[47]. Мы использовали Т-инвариантный анализ для проверки надежности модели. Всего было вычислено 393 Т-инварианта плюс 10 тривиальных инвариантов, которые были исключены из анализа. Они были сгруппированы в 44 кластера, а с помощью Т-инвариантов переходы были сгруппированы в 40 MCTS. Полученная модель связна, покрыта Т-инвариантами, каждый из которых имеет биологический смысл.Анализ MCTS использовался для определения наиболее частых критических переходов, происходящих в модели. Это конкретное подмножество в дальнейшем использовалось для планирования экспериментов по выбиванию in silico , а также для проверки модели и анализа ожидаемого биологического поведения. Затем модель была использована для выполнения in silico экспериментов по отключению определенных переходов во время качественного сетевого анализа. Наши результаты показали, что модель способна представлять важные переходы, отражающие реальные биологические результаты, т.е.е. переходы с участием таких соединений, как кислород или АТФ, правильно инактивируются при определенных обстоятельствах, как и ожидалось из библиографических данных.
Мы использовали Т-инвариантный анализ для проверки надежности модели. Всего было вычислено 393 Т-инварианта плюс 10 тривиальных инвариантов, которые были исключены из анализа. Они были сгруппированы в 44 кластера, а с помощью Т-инвариантов переходы были сгруппированы в 40 MCTS. Полученная модель связна, покрыта Т-инвариантами, каждый из которых имеет биологический смысл.Анализ MCTS использовался для определения наиболее частых критических переходов, происходящих в модели. Это конкретное подмножество в дальнейшем использовалось для планирования экспериментов по выбиванию in silico , а также для проверки модели и анализа ожидаемого биологического поведения. Затем модель была использована для выполнения in silico экспериментов по отключению определенных переходов во время качественного сетевого анализа. Наши результаты показали, что модель способна представлять важные переходы, отражающие реальные биологические результаты, т.е.е. переходы с участием таких соединений, как кислород или АТФ, правильно инактивируются при определенных обстоятельствах, как и ожидалось из библиографических данных.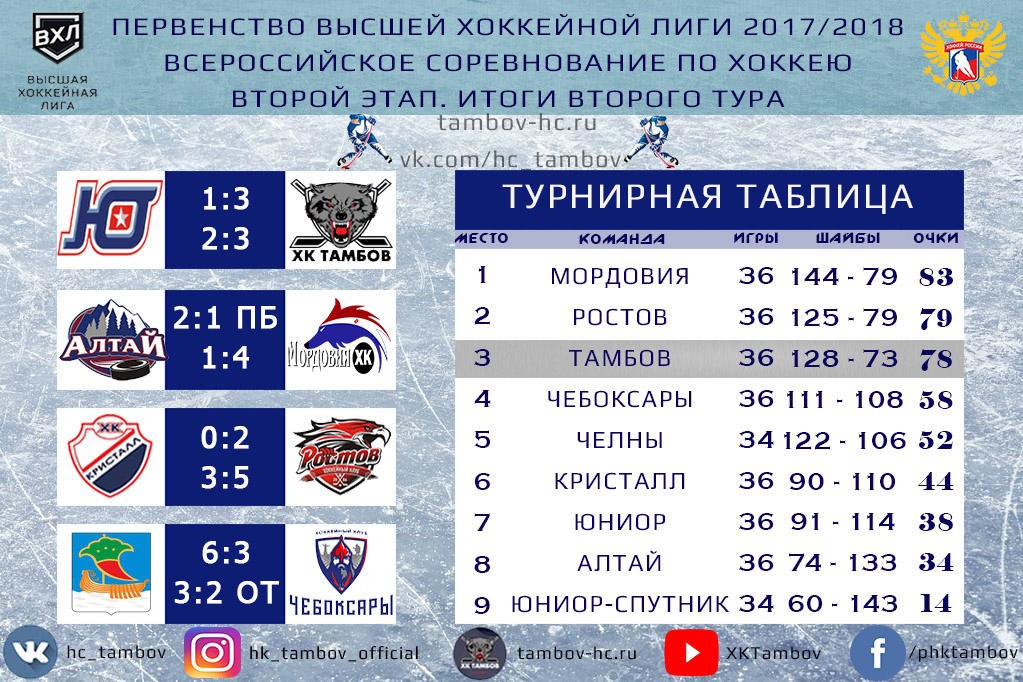 Биологические энергетические реакции (например, образование АТФ из АДФ) моделировались как P-инварианты. Хотя сеть намеренно не покрыта P-инвариантами, P-инвариантный анализ использовался для проверки всех смоделированных переходов с потреблением энергии. Балансы АТФ и НАДН казались постоянными во время моделирования, с нерелевантными P-инвариантами, расположенными в системе Hur.Это приближение было использовано для проверки Hur-зависимой регуляции VEGF с результатами в соответствии с [50]. Специфический нокаут pVHL предполагает, что одного этого белка недостаточно для полной инактивации HIF-1α. Действительно, другие параллельные пути деградации HIF-1α способствуют своего рода резервной регуляции клеточного цикла. Напротив, оказалось, что простой делеции pVHL достаточно, чтобы усилить все другие ее ингибирующие функции, проявляя сходные эффекты с патологическими симптомами VHL. Действительно, дестабилизация ВКМ увеличивает миграцию клеток в другие области, способствуя возникновению метастазов в случае опухолевых клеток.
Биологические энергетические реакции (например, образование АТФ из АДФ) моделировались как P-инварианты. Хотя сеть намеренно не покрыта P-инвариантами, P-инвариантный анализ использовался для проверки всех смоделированных переходов с потреблением энергии. Балансы АТФ и НАДН казались постоянными во время моделирования, с нерелевантными P-инвариантами, расположенными в системе Hur.Это приближение было использовано для проверки Hur-зависимой регуляции VEGF с результатами в соответствии с [50]. Специфический нокаут pVHL предполагает, что одного этого белка недостаточно для полной инактивации HIF-1α. Действительно, другие параллельные пути деградации HIF-1α способствуют своего рода резервной регуляции клеточного цикла. Напротив, оказалось, что простой делеции pVHL достаточно, чтобы усилить все другие ее ингибирующие функции, проявляя сходные эффекты с патологическими симптомами VHL. Действительно, дестабилизация ВКМ увеличивает миграцию клеток в другие области, способствуя возникновению метастазов в случае опухолевых клеток. Кроме того, зависимое от pVHL ингибирование образования плотных контактов с помощью aPKCζII участвует в более легком отслоении клеток. Взаимодействия Nur77 можно считать хорошим примером патологических эффектов. Является стимулятором выработки проопиомеланокортина, предшественника адренокортикотропного гормона. При чрезмерном высвобождении он способствует перепроизводству адренергических нейротрансмиттеров надпочечниками. Приближается к клиническому состоянию, известному как синдром Кушинга. Известно, что в долгосрочной перспективе дерегуляция Nur77 вызывает опухоли гипофиза и надпочечников [51], [52].Это происходит при феохромоцитоме, которая является одним из основных проявлений болезни ВХЛ. Мы предполагаем, что непрерывная транскрипция VEGF, даже в ситуациях, когда HIF-1α (но не Sp1) нокаутирован, может быть объяснением клинических исследований, в которых препараты, направленные на VEGF, оказались эффективными при лечении рака почки, как сообщалось в [53]. ]. Хотя мы использовали только подтвержденные литературные данные, Nur77 может быть вовлечен в другие системы регуляции, которые не учитывались в нашей модели.
Кроме того, зависимое от pVHL ингибирование образования плотных контактов с помощью aPKCζII участвует в более легком отслоении клеток. Взаимодействия Nur77 можно считать хорошим примером патологических эффектов. Является стимулятором выработки проопиомеланокортина, предшественника адренокортикотропного гормона. При чрезмерном высвобождении он способствует перепроизводству адренергических нейротрансмиттеров надпочечниками. Приближается к клиническому состоянию, известному как синдром Кушинга. Известно, что в долгосрочной перспективе дерегуляция Nur77 вызывает опухоли гипофиза и надпочечников [51], [52].Это происходит при феохромоцитоме, которая является одним из основных проявлений болезни ВХЛ. Мы предполагаем, что непрерывная транскрипция VEGF, даже в ситуациях, когда HIF-1α (но не Sp1) нокаутирован, может быть объяснением клинических исследований, в которых препараты, направленные на VEGF, оказались эффективными при лечении рака почки, как сообщалось в [53]. ]. Хотя мы использовали только подтвержденные литературные данные, Nur77 может быть вовлечен в другие системы регуляции, которые не учитывались в нашей модели. Переходы для стабилизации фибронектина pVHL показывают поведение, которое согласуется с биохимическими экспериментами, иллюстрируя полную отмену стабилизации ECM и повышенное действие матриксной металлопротеиназы.Хотя результаты обнадеживают, представленная модель нуждается в дальнейших улучшениях, поскольку стандартные PN не обеспечивают ни полного контроля перехода, ни модуляции ферментативной активности. Тем не менее, благодаря ручному курированию нашу модель можно использовать для планирования новых экспериментов in vitro и in vivo . Результаты достаточно убедительны, чтобы предложить нашу модель в качестве комплексной модели пути для моделирования основных функций pVHL.
Переходы для стабилизации фибронектина pVHL показывают поведение, которое согласуется с биохимическими экспериментами, иллюстрируя полную отмену стабилизации ECM и повышенное действие матриксной металлопротеиназы.Хотя результаты обнадеживают, представленная модель нуждается в дальнейших улучшениях, поскольку стандартные PN не обеспечивают ни полного контроля перехода, ни модуляции ферментативной активности. Тем не менее, благодаря ручному курированию нашу модель можно использовать для планирования новых экспериментов in vitro и in vivo . Результаты достаточно убедительны, чтобы предложить нашу модель в качестве комплексной модели пути для моделирования основных функций pVHL.
Мутация гена VHL типа 2B снижает дозировку HIF in vitro и in vivo
Alvarez-Silva M, Belo-Diabangouaya P, Salaun J, Dieterlen-Lievre F .(2003). Плацента мыши является основным органом кроветворения. Разработка 130 : 5437–5444.
КАС Статья пабмед Google Scholar
Анг С.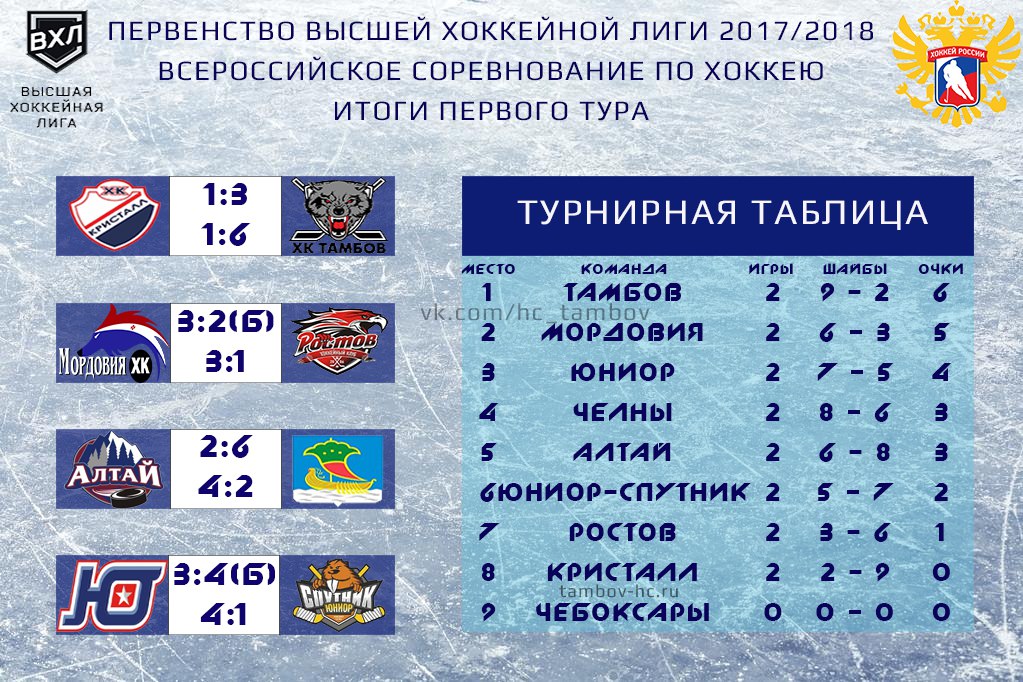 О., Чен Х., Хирота К., Гордеук В.Р., Елинек Дж., Гуан И. и др. . (2002). В основе врожденной чувашской полицитемии лежит нарушение кислородного гомеостаза. Нат Жене 32 : 614–621.
О., Чен Х., Хирота К., Гордеук В.Р., Елинек Дж., Гуан И. и др. . (2002). В основе врожденной чувашской полицитемии лежит нарушение кислородного гомеостаза. Нат Жене 32 : 614–621.
КАС Статья пабмед Google Scholar
Бэнкс Р.Е., Тируконда П., Тейлор С., Хорниголд Н., Астути Д., Коэн Д. и др. .(2006). Генетический и эпигенетический анализ изменений гена фон Хиппеля-Линдау (VHL) и взаимосвязи с клиническими переменными при спорадическом раке почки. Рак Res 66 : 2000–2011.
КАС Статья пабмед Google Scholar
Брюик Р.К., Макнайт С.Л. (2001). Консервативное семейство пролил-4-гидроксилаз, модифицирующих HIF. Наука 294 : 1337–1340.
КАС Статья пабмед Google Scholar
Брюссельманс К., Боно Ф., Коллен Д. , Герберт Дж.М., Кармели П., Деверчин М. .(2005). Новая роль сосудистого эндотелиального фактора роста как аутокринного фактора выживания эмбриональных стволовых клеток во время гипоксии. J Biol Chem 280 : 3493–3499.
, Герберт Дж.М., Кармели П., Деверчин М. .(2005). Новая роль сосудистого эндотелиального фактора роста как аутокринного фактора выживания эмбриональных стволовых клеток во время гипоксии. J Biol Chem 280 : 3493–3499.
КАС Статья пабмед Google Scholar
Кармели П., Дор Ю., Герберт Дж. М., Фукумура Д., Брюссельманс К., Деверчин М. и др. . (1998). Роль HIF-1альфа в опосредованном гипоксией апоптозе, пролиферации клеток и опухолевом ангиогенезе. Природа 394 : 485–490.
КАС Статья пабмед Google Scholar
Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH и др. . (2001). Контрастные эффекты на регуляцию HIF-1альфа вызывающими заболевание мутациями pVHL коррелируют с моделями онкогенеза при болезни фон Гиппеля-Линдау. Hum Mol Genet 10 : 1029–1038.
КАС Статья пабмед Google Scholar
Кокман М. Е., Массон Н., Моул Д.Р., Яаккола П., Чанг Г.В., Клиффорд С.К. и др. .(2000). Индуцируемое гипоксией связывание фактора-альфа и убиквитилирование белком-супрессором опухоли фон Хиппеля-Линдау. J Biol Chem 275 : 25733–25741.
Е., Массон Н., Моул Д.Р., Яаккола П., Чанг Г.В., Клиффорд С.К. и др. .(2000). Индуцируемое гипоксией связывание фактора-альфа и убиквитилирование белком-супрессором опухоли фон Хиппеля-Линдау. J Biol Chem 275 : 25733–25741.
КАС Статья пабмед Google Scholar
Ковелло К.Л., Саймон М.К., Кит Б. (2005). Целенаправленная замена индуцируемого гипоксией фактора-1альфа нокаут-аллелем индуцируемого гипоксией фактора-2альфа способствует росту опухоли. Рак Res 65 : 2277–2286.
КАС Статья пабмед Google Scholar
Кросси П.А., Ричардс Ф.М., Фостер К., Грин Дж.С., Проуз А., Латиф Ф. и др. . (1994). Выявление внутригенных мутаций в гене-супрессоре опухоли при болезни фон Хиппеля-Линдау и корреляция с фенотипом заболевания. Hum Mol Genet 3 : 1303–1308.
КАС Статья пабмед Google Scholar
Дуан Д. Р., Пауза А., Берджесс В.Х., Асо Т., Чен Д.Ю., Гарретт К.П. и др. .(1995). Ингибирование элонгации транскрипции белком-супрессором опухоли VHL. Наука 269 : 1402–1406.
Р., Пауза А., Берджесс В.Х., Асо Т., Чен Д.Ю., Гарретт К.П. и др. .(1995). Ингибирование элонгации транскрипции белком-супрессором опухоли VHL. Наука 269 : 1402–1406.
КАС Статья пабмед Google Scholar
Элверт Г., Каппель А., Хайденрайх Р., Энглмайер Ю., Ланц С., Акер Т. и др. . (2003). Кооперативное взаимодействие индуцируемого гипоксией фактора-2альфа (HIF-2альфа) и Ets-1 при активации транскрипции рецептора-2 фактора роста эндотелия сосудов (Flk-1). J Biol Chem 278 : 7520–7530.
КАС Статья пабмед Google Scholar
Эпштейн А.С., Глидл Дж.М., Макнейл Л.А., Хьюитсон К.С., О’Рурк Дж., Моул Д.Р. и др. . (2001). C. elegans EGL-9 и гомологи млекопитающих определяют семейство диоксигеназ, которые регулируют HIF путем гидроксилирования пролила. Сотовый 107 : 43–54.
КАС Статья пабмед Google Scholar
Фостер К. , Проуз А., ван ден Берг А., Флеминг С., Халсбик М.М., Кросси П.А. и др. .(1994). Соматические мутации гена-супрессора опухоли фон Хиппеля-Линдау при несемейной светлоклеточной карциноме почки. Hum Mol Genet 3 : 2169–2173.
, Проуз А., ван ден Берг А., Флеминг С., Халсбик М.М., Кросси П.А. и др. .(1994). Соматические мутации гена-супрессора опухоли фон Хиппеля-Линдау при несемейной светлоклеточной карциноме почки. Hum Mol Genet 3 : 2169–2173.
КАС Статья пабмед Google Scholar
Gallou C, Chauveau D, Richard S, Joly D, Giraud S, Olschwang S и др. . (2004). Корреляция генотип-фенотип в семьях фон Хиппеля-Линдау с поражением почек. Хум Мутат 24 : 215–224.
КАС Статья пабмед Google Scholar
Гнарра Дж. Р., Уорд Дж. М., Портер Ф. Д., Вагнер Дж. Р., Девор Д. Е., Гринберг А. и др. . (1997). Дефектный плацентарный васкулогенез вызывает эмбриональную летальность у мышей с дефицитом VHL. Proc Natl Acad Sci USA 94 : 9102–9107.
КАС Статья пабмед Google Scholar
Хаазе В.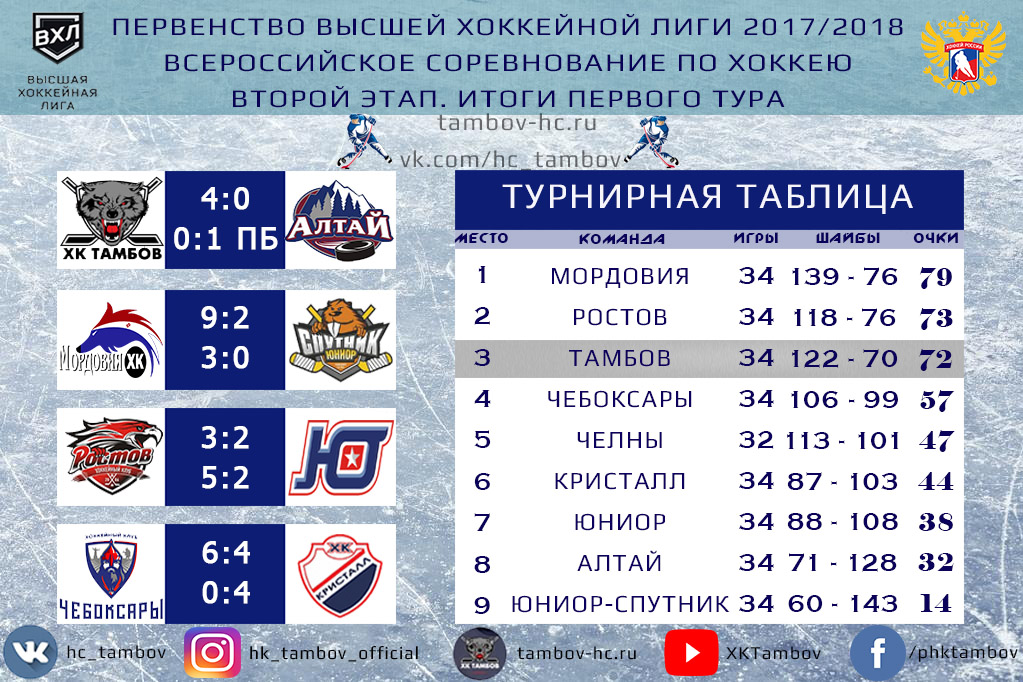 Х., Гликман Ю.Н., Соколовский М., Йениш Р. .(2001). Сосудистые опухоли печени с направленной инактивацией опухолевого супрессора фон Хиппеля-Линдау. Proc Natl Acad Sci USA 98 : 1583–1588.
Х., Гликман Ю.Н., Соколовский М., Йениш Р. .(2001). Сосудистые опухоли печени с направленной инактивацией опухолевого супрессора фон Хиппеля-Линдау. Proc Natl Acad Sci USA 98 : 1583–1588.
КАС Статья пабмед ПабМед Центральный Google Scholar
Хакер К.Е., Ли К.М., Ратмелл В.К. (2008). Мутации VHL типа 2B сохраняют сложную форму и функцию VBC. PLoS One 3 : e3801.
Артикул пабмед ПабМед Центральный Google Scholar
Хики М.М., Лэм Дж. К., Безман Н. А., Ратмелл В. К., Саймон М. С. .(2007). Мутация фон Хиппеля-Линдау у мышей повторяет чувашскую полицитемию посредством индуцируемой гипоксией передачи сигналов фактора-2альфа и эритропоэза в селезенке. J Clin Invest 117 : 3879–3889.
КАС пабмед ПабМед Центральный Google Scholar
Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW и др.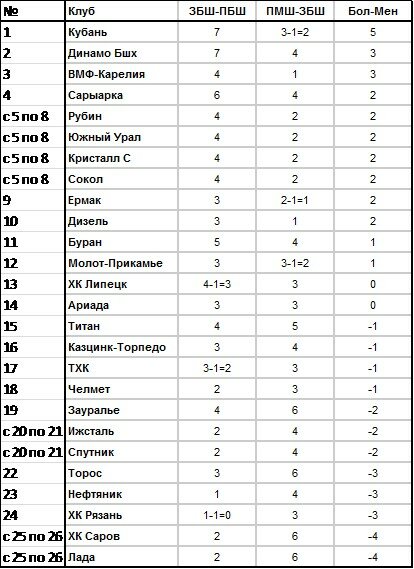 . (2002). Структурная основа распознавания гидроксипролина в HIF-1 альфа с помощью pVHL. Природа 417 : 975–978.
. (2002). Структурная основа распознавания гидроксипролина в HIF-1 альфа с помощью pVHL. Природа 417 : 975–978.
КАС Статья пабмед Google Scholar
Hu CJ, Iyer S, Sataur A, Covello KL, Chodosh LA, Simon MC . (2006). Дифференциальная регуляция транскрипционной активности индуцируемого гипоксией фактора 1 альфа (HIF-1альфа) и HIF-2альфа в стволовых клетках. Mol Cell Biol 26 : 3514–3526.
КАС Статья пабмед ПабМед Центральный Google Scholar
Ху С.Дж., Ван Л.И., Ходош Л.А., Кит Б., Саймон М.С.(2003). Различная роль индуцируемого гипоксией фактора 1альфа (HIF-1альфа) и HIF-2альфа в регуляции генов гипоксии. Mol Cell Biol 23 : 9361–9374.
КАС Статья пабмед ПабМед Центральный Google Scholar
Хуан Дж., Чжао К. , Муни С.М., Ли Ф.С. (2002). Детерминанты последовательности индуцируемого гипоксией фактора-1альфа для гидроксилирования пролилгидроксилазами PHD1, PHD2 и PHD3. J Biol Chem 277 : 39792–39800.
, Муни С.М., Ли Ф.С. (2002). Детерминанты последовательности индуцируемого гипоксией фактора-1альфа для гидроксилирования пролилгидроксилазами PHD1, PHD2 и PHD3. J Biol Chem 277 : 39792–39800.
КАС Статья пабмед ПабМед Центральный Google Scholar
Хуанг Л.Е., Гу Дж., Шау М., Банн Х.Ф. (1998). Регуляция индуцируемого гипоксией фактора 1альфа опосредуется О2-зависимым доменом деградации через убиквитин-протеасомный путь. Proc Natl Acad Sci USA 95 : 7987–7992.
КАС Статья пабмед Google Scholar
Илиопулос О., Кибел А., Грей С., Келин-младший WG .(1995). Подавление опухоли человеческим продуктом гена фон Хиппеля-Линдау. Nat Med 1 : 822–826.
КАС Статья пабмед Google Scholar
Илиопулос О., Леви А.П., Цзян С., Кэлин младший В.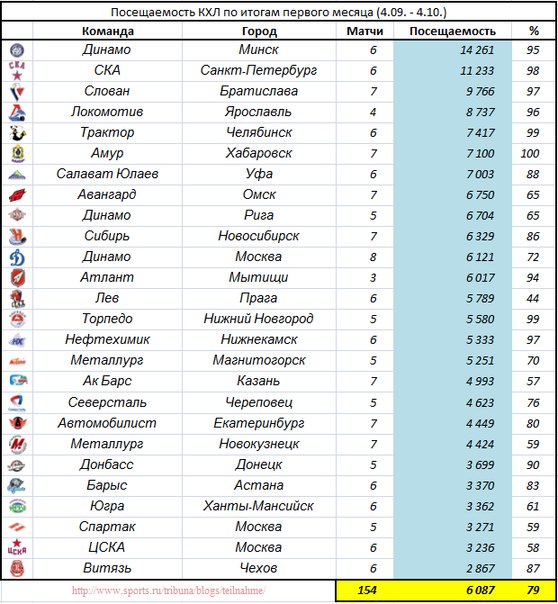 Г., Голдберг М.А. (1996). Негативная регуляция генов, индуцируемых гипоксией, белком фон Хиппеля-Линдау. Proc Natl Acad Sci USA 93 : 10595–10599.
Г., Голдберг М.А. (1996). Негативная регуляция генов, индуцируемых гипоксией, белком фон Хиппеля-Линдау. Proc Natl Acad Sci USA 93 : 10595–10599.
КАС Статья пабмед Google Scholar
Иван М., Хабербергер Т., Герваси Д.С., Михельсон К.С., Гюнцлер В., Кондо К. и др. .(2002). Биохимическая очистка и фармакологическое ингибирование пролилгидроксилазы млекопитающих, действующей на фактор, индуцируемый гипоксией. Proc Natl Acad Sci USA 99 : 13459–13464.
КАС Статья пабмед Google Scholar
Иван М., Кондо К., Ян Х., Ким В., Валиандо Дж., Ох М. и др. . (2001). HIF-альфа, нацеленный на VHL-опосредованное разрушение путем гидроксилирования пролина: последствия для восприятия O2. Наука 292 : 464–468.
КАС Статья пабмед ПабМед Центральный Google Scholar
Иваи К. , Яманака К., Камура Т., Минато Н., Конэуэй Р.С., Конэвей Дж.В. и др. . (1999). Идентификация белка-супрессора опухоли фон Хиппель-Линдау как части активного комплекса убиквитинлигазы E3. Proc Natl Acad Sci USA 96 : 12436–12441.
, Яманака К., Камура Т., Минато Н., Конэуэй Р.С., Конэвей Дж.В. и др. . (1999). Идентификация белка-супрессора опухоли фон Хиппель-Линдау как части активного комплекса убиквитинлигазы E3. Proc Natl Acad Sci USA 96 : 12436–12441.
КАС Статья пабмед Google Scholar
Яаккола П., Моул Д.Р., Тиан Ю.М., Уилсон М.И., Гилберт Дж., Гаскелл С.Дж. и др. .(2001). Нацеливание HIF-альфа на комплекс убиквитилирования фон Хиппеля-Линдау путем гидроксилирования пролила, регулируемого O2. Наука 292 : 468–472.
КАС Статья пабмед ПабМед Центральный Google Scholar
Цзян Б.Х., Рю Э., Ван Г.Л., Роу Р., Семенца Г.Л. (1996). Свойства димеризации, связывания ДНК и трансактивации фактора 1, индуцируемого гипоксией. J Biol Chem 271 : 17771–17778.
КАС Статья пабмед Google Scholar
Цзян Б. Х., Чжэн Дж.З., Люн С.В., Роу Р., Семенца Г.Л. (1997). Трансактивационные и ингибирующие домены индуцируемого гипоксией фактора 1альфа. Модуляция транскрипционной активности напряжением кислорода. J Biol Chem 272 : 19253–19260.
Х., Чжэн Дж.З., Люн С.В., Роу Р., Семенца Г.Л. (1997). Трансактивационные и ингибирующие домены индуцируемого гипоксией фактора 1альфа. Модуляция транскрипционной активности напряжением кислорода. J Biol Chem 272 : 19253–19260.
КАС Статья Google Scholar
Камура Т., Кепп Д.М., Конрад М.Н., Сковыра Д., Морленд Р.Дж., Илиопулос О. и др. .(1999). Rbx1, компонент комплекса супрессора опухоли VHL и убиквитинлигазы SCF. Наука 284 : 657–661.
КАС Статья пабмед ПабМед Центральный Google Scholar
Камура Т., Сато С., Иваи К., Чижик-Кшеска М., Конэуэй Р.С., Конэуэй Дж.В. (2000). Активация убиквитинирования HIF1-альфа восстановленным комплексом опухолевых супрессоров фон Хиппеля-Линдау (VHL). Proc Natl Acad Sci USA 97 : 10430–10435.
КАС Статья пабмед ПабМед Центральный Google Scholar
Кибель А. , Илиопулос О., Де Каприо Дж. А., Келин младший РГ. (1995). Связывание белка-супрессора опухоли фон Хиппеля-Линдау с элонгинами B и C. Science 269 : 1444–1446.
, Илиопулос О., Де Каприо Дж. А., Келин младший РГ. (1995). Связывание белка-супрессора опухоли фон Хиппеля-Линдау с элонгинами B и C. Science 269 : 1444–1446.
КАС Статья пабмед Google Scholar
Кнаут К., Бекс С., Джемт П., Бухбергер А. .(2006). Риск почечно-клеточной карциномы при болезни Гиппеля-Линдау 2 типа коррелирует с дефектами стабильности pVHL и взаимодействиями HIF-1альфа. Онкоген 25 : 370–377.
КАС Статья пабмед Google Scholar
Кобаяши Т., Минова О., Куно Дж., Митани Х., Хино О., Нода Т. . (1999). Почечный канцерогенез, гемангиоматоз печени и эмбриональная летальность, вызванная мутацией Tsc2 зародышевой линии у мышей. Рак Res 59 : 1206–1211.
КАС пабмед Google Scholar
Кондо К., Яо М., Йошида М., Кисида Т.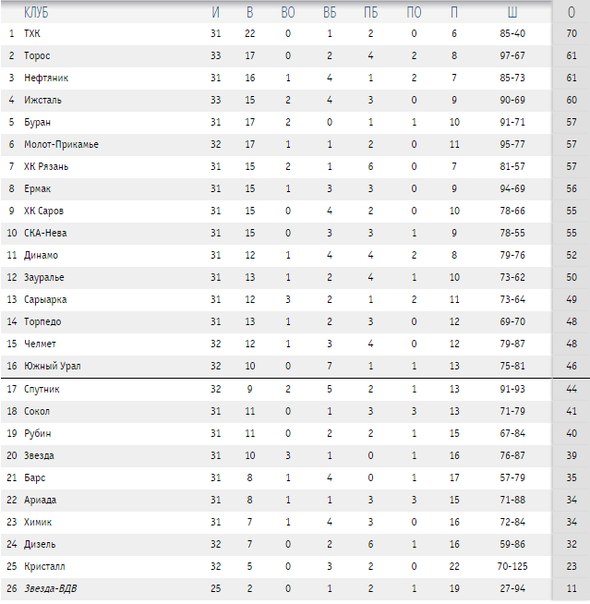 , Шуин Т., Миура Т. и др. . (2002). Комплексный мутационный анализ гена VHL при спорадической почечно-клеточной карциноме: связь с клинико-патологическими параметрами. Гены Хромосомы Рак 34 : 58–68.
, Шуин Т., Миура Т. и др. . (2002). Комплексный мутационный анализ гена VHL при спорадической почечно-клеточной карциноме: связь с клинико-патологическими параметрами. Гены Хромосомы Рак 34 : 58–68.
КАС Статья пабмед Google Scholar
Латиф Ф., Тори К., Гнарра Дж., Яо М., Дах Ф.М., Оркатт М.Л. и др. .(1993). Идентификация гена-супрессора опухоли при болезни фон Гиппеля-Линдау. Наука 260 : 1317–1320.
КАС Статья пабмед ПабМед Центральный Google Scholar
Ле Бра А, Лайоннетон Ф, Матто В, Лелиевре Э, Каэтано Б, Спрюйт Н и др. . (2007). HIF-2-альфа специфически активирует промотор VE-кадгерина независимо от гипоксии и в синергии с Ets-1 через два основных сайта связывания ETS. Онкоген 26 : 7480–7489.
КАС Статья пабмед Google Scholar
Ли Л. , Чжан Л., Чжан С., Ян К., Минамисима Ю.А., Олуми А.Ф. и др. . (2007). Фактор, индуцируемый гипоксией, связан с дифференциальным риском рака почки, наблюдаемым при мутациях VHL типа 2A и типа 2B. Mol Cell Biol 27 : 5381–5392.
, Чжан Л., Чжан С., Ян К., Минамисима Ю.А., Олуми А.Ф. и др. . (2007). Фактор, индуцируемый гипоксией, связан с дифференциальным риском рака почки, наблюдаемым при мутациях VHL типа 2A и типа 2B. Mol Cell Biol 27 : 5381–5392.
КАС Статья пабмед ПабМед Центральный Google Scholar
Листван Дж., Имберт Г., Вирбелауэр С., Гстайгер М., Крек В.. (1999). Белок-супрессор опухоли фон Хиппеля-Линдау является компонентом активности убиквитин-протеинлигазы Е3. Гены Дев 13 : 1822–1833.
КАС Статья пабмед ПабМед Центральный Google Scholar
Ма В., Тессаролло Л., Хонг С.Б., Баба М., Саутон Э., Бэк ТС и др. . (2003). Опухоли сосудов печени, ангиоэктазии во многих органах и нарушение сперматогенеза у мышей с условной инактивацией гена VHL. Рак Res 63 : 5320–5328.
КАС пабмед Google Scholar
Мак Ф. А., Ратмелл В.К., Аршам А.М., Гнарра Дж., Кит Б., Саймон М.К. (2003). Потеря pVHL достаточна, чтобы вызвать дисрегуляцию HIF в первичных клетках, но не способствует росту опухоли. Раковая клетка 3 : 75–88.
А., Ратмелл В.К., Аршам А.М., Гнарра Дж., Кит Б., Саймон М.К. (2003). Потеря pVHL достаточна, чтобы вызвать дисрегуляцию HIF в первичных клетках, но не способствует росту опухоли. Раковая клетка 3 : 75–88.
КАС Статья пабмед ПабМед Центральный Google Scholar
Махер Э.Р., Иселиус Л., Йейтс Дж.Р., Литтлер М., Бенджамин С., Харрис Р. и др. .(1991). Болезнь фон Гиппеля-Линдау: генетическое исследование. J Med Genet 28 : 443–447.
КАС Статья пабмед ПабМед Центральный Google Scholar
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME и др. . (1999). Белок-супрессор опухоли VHL нацелен на факторы, индуцируемые гипоксией, для кислородзависимого протеолиза. Природа 399 : 271–275.
КАС Статья пабмед ПабМед Центральный Google Scholar
Миллер М. С. (2004). Трансплацентарный канцерогенез легких: молекулярные механизмы и патогенез. Toxicol Appl Pharmacol 198 : 95–110.
С. (2004). Трансплацентарный канцерогенез легких: молекулярные механизмы и патогенез. Toxicol Appl Pharmacol 198 : 95–110.
КАС Статья пабмед Google Scholar
Мин Дж.Х., Ян Х., Иван М., Гертлер Ф., Келин младший В.Г., Павлетич Н.П. (2002).Структура комплекса HIF-1альфа-pVHL: распознавание гидроксипролина в передаче сигналов. Наука 296 : 1886–1889.
КАС Статья пабмед Google Scholar
Neumann HP, Wiestler OD . (1991). Кластеризация признаков синдрома фон Хиппеля-Линдау: свидетельство сложного генетического локуса. Ланцет 337 : 1052–1054.
КАС Статья пабмед Google Scholar
Никерсон М.Л., Джагер Э., Ши Ю., Дурочер Дж.А., Махуркар С., Заридзе Д. и др. .(2008). Улучшенная идентификация изменений гена фон Хиппеля-Линдау в светлоклеточных опухолях почек.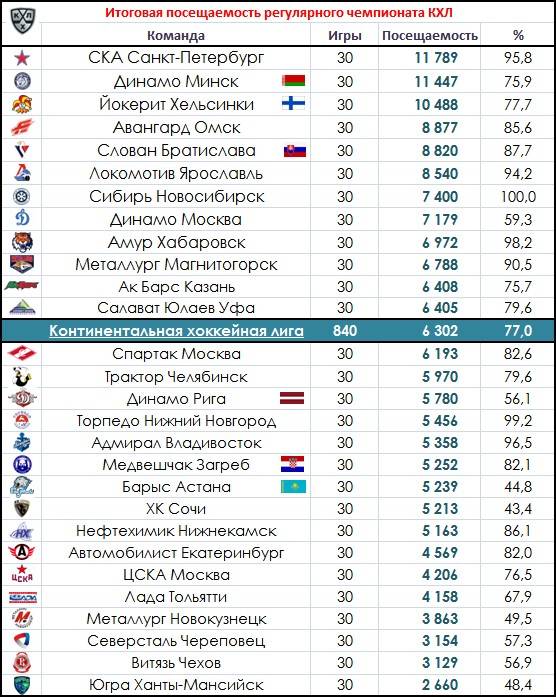 Clin Cancer Res 14 : 4726–4734.
Clin Cancer Res 14 : 4726–4734.
КАС Статья пабмед ПабМед Центральный Google Scholar
Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE и др. . (2000). Убиквитинирование фактора, индуцируемого гипоксией, требует прямого связывания с бета-доменом белка фон Хиппеля-Линдау. Nat Cell Biol 2 : 423–427.
КАС Статья пабмед Google Scholar
Ох М., Такаги Ю., Асо Т., Стеббинс С.Е., Павлетич Н.П., Збар Б. и др. . (1999). Синтетические пептиды определяют критические контакты между элонгином С, элонгином В и белком фон Хиппеля-Линдау. J Clin Invest 104 : 1583–1591.
КАС Статья пабмед ПабМед Центральный Google Scholar
Пасторе Ю.Д., Елинек Дж., Анг С., Гуан Ю., Лю Э., Джедличкова К. и др. . (2003). Мутации в гене VHL при спорадической, по-видимому, врожденной полицитемии. Кровь 101 : 1591–1595.
(2003). Мутации в гене VHL при спорадической, по-видимому, врожденной полицитемии. Кровь 101 : 1591–1595.
КАС Статья пабмед Google Scholar
Пауза А., Ли С., Уоррелл Р.А., Чен Д.Ю., Берджесс В.Х., Линехан В.М. и др. . (1997). Продукт гена-супрессора опухоли фон Хиппеля-Линдау образует стабильный комплекс с человеческим CUL-2, членом семейства белков Cdc53. Proc Natl Acad Sci USA 94 : 2156–2161.
КАС Статья пабмед Google Scholar
Пью К.В., О’Рурк Дж.Ф., Нагао М., Глидл Дж.М., Рэтклифф П.Дж. (1997). Активация гипоксия-индуцируемого фактора-1; определение регуляторных доменов внутри альфа-субъединицы. J Biol Chem 272 : 11205–11214.
КАС Статья пабмед Google Scholar
Ратмелл В.К., Чен С. .(2008). Инактивация VHL при почечно-клеточном раке: значение для диагностики, прогноза и лечения. Expert Rev Anticancer Ther 8 : 63–73.
Expert Rev Anticancer Ther 8 : 63–73.
КАС Статья пабмед ПабМед Центральный Google Scholar
Rathmell WK, Hickey MM, Bezman NA, Chmielecki CA, Carraway NC, Simon MC . (2004). In vitro и in vivo модели анализируют мутации, специфичные для болезни фон Хиппеля-Линдау. Рак Res 64 : 8595–8603.
КАС Статья пабмед Google Scholar
Санчес-Эльснер Т., Ботелла Л.М., Веласко Б., Ланга К., Бернабеу К. . (2002). Экспрессия эндоглина регулируется транскрипционным сотрудничеством между путями гипоксии и трансформирующего фактора роста-бета. J Biol Chem 277 : 43799–43808.
КАС Статья пабмед Google Scholar
Семенза Г.Л.(2003). Ориентация на HIF-1 для лечения рака. Nat Rev Рак 3 : 721–732.
КАС Статья пабмед ПабМед Центральный Google Scholar
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P и др. . (1996). Элементы реакции на гипоксию в промоторах генов альдолазы А, енолазы 1 и лактатдегидрогеназы А содержат важные сайты связывания фактора 1, индуцируемого гипоксией. J Biol Chem 271 : 32529–32537.
КАС Статья пабмед Google Scholar
Стеббинс С.Е., Каэлин младший В.Г., Павлетич Н.П. (1999). Структура комплекса VHL-ElonginC-ElonginB: влияние на супрессорную функцию VHL. Наука 284 : 455–461.
КАС Статья пабмед Google Scholar
Тиан Х., Макнайт С.Л., Рассел Д.В.(1997). Эндотелиальный белок домена PAS 1 (EPAS1), фактор транскрипции, селективно экспрессируемый в эндотелиальных клетках. Гены Дев 11 : 72–82.
КАС Статья пабмед Google Scholar
Восс А.К., Томас Т., Грусс П. . (2000). У мышей, лишенных HSP90beta, не развивается плацентарный лабиринт. Разработка 127 : 1–11.
КАС пабмед ПабМед Центральный Google Scholar
Williams-Simons L, Westphal H .(1999). EIIaCre — полезность штамма общего делетора. Transgenic Res 8 : 53–54.
КАС Статья пабмед Google Scholar
Збар Б., Кишида Т., Чен Ф., Шмидт Л., Махер Э.Р., Ричардс Ф.М. и др. . (1996). Мутации зародышевой линии в гене болезни фон Хиппеля-Линдау (VHL) в семьях из Северной Америки, Европы и Японии. Хум Мутат 8 : 348–357.
КАС Статья пабмед Google Scholar
Изменения гена VHL у пациентов с почечно-клеточным раком: новые очаговые мутации или мутации-основатели и неравновесное сцепление
Beroud C, Joly D, Gallou C, Staroz F, Orfanelli MT, Junien C . 1998 Nucleic Acids Res. 26 : 256–258
1998 Nucleic Acids Res. 26 : 256–258
Браух Х., Вейрих Г., Бригер Дж., Главац Д., Родл Х., Айхингер М., Фойрер М., Вайдт Э., Пуранаканиттха К., Нейхаус К., Помер С., Бреннер В., Ширмахер П. , Сторкель С., Роттер М., Масера А., Гугелер Н., Декер Х.Дж. 2000 Рак Рез. 60 : 1942–1948
Браух Х., Вейрих Г., Хорнауэр М.А., Шторкель С., Воль Т., Брюнинг Т. . 1999 J. Natl. Рак инст. 91 : 854–861
Брюнинг Т., Вейрих Г., Хорнауэр М.А., Хофлер Х., Браух Х. .1997 Арх. Токсикол. 71 : 332–335
Кросси П.А., Ричардс Ф.М., Фостер К., Грин Дж.С., Проуз А., Латиф Ф., Лерман М.И., Збар Б., Аффара Н.А., Фергюсон-Смит М.А. 1994 Гул. Мол. Жене 3 : 1303–1308
Дэй РСД, Бабич М.А., Ярош Д.Б., Скудиеро Д.А. 1987 J. Cell Sci. Suppl 6 : 333–353
Денисенко М.Ф., Чен Д.С., Тан М.С., Пфайфер Г.П. 1997 Проц. Натл. акад.науч. США 94 : 3893–3898
Натл. акад.науч. США 94 : 3893–3898
Фостер К., Проуз А., ван ден Берг А., Флеминг С., Халсбек М.М., Кросси П.А., Ричардс Ф.М., Кэрнс П., Аффара Н.А., Фергюсон-Смит М.А., Buys CHCM, Махер ЭР. 1994 Гул. Мол. Жене. 3 : 2169–2173
Гнарра Дж.Р., Тори К., Венг Ю., Шмидт Л., Вей М.Х., Ли Х., Латиф Ф., Лю С., Чен Ф., Дух Ф.М., Лубенский И., Дуан Д.Р., Флоренс С. , Pozzatti R, Walther MM, Bander NH, Grossman HB, Brauch H, Pomer S, Brooks WB, Isaacs WB, Lehman MI, Zbar B, Linehan WM .1994 Нац. Жене. 7 : 85–90
Гороспе М., Иган Дж. М., Збар Б., Лерман М., Гейл Л., Кузьмин И., Холбрук Н. Дж. . 1999 Мол. Клеточная биол. 19 : 1289–1300
Гринблатт М.С., Беннетт В.П., Холлштейн М., Харрис К.С. 1994 Рак Рез. 54 : 4855–4878
Латиф Ф., Тори К., Гнарра Дж., Яо М., Дах Ф.М., Оркатт М.Л., Стакхаус Т., Кузьмин И., Моди В., Гейл Л. , Шмидт Л., Чжоу Ф., Ли Х. , Вэй М.Х., Чен Ф., Гленн Г., Чойке П., Макклеллан М.В., Венг Ю., Дуан Д-СР, Дин М., Главац Д., Ричардс Ф.М., Кросси П.А., Фергюсон-Смит М.А., Ле Паслье Д., Чумаков И., Коэн Д. , Чинаулт А.Г., Махер Э.Р., Линехан В.М., Збар Б., Лерман М.И.1993 Наука 260 : 1317–1320
, Шмидт Л., Чжоу Ф., Ли Х. , Вэй М.Х., Чен Ф., Гленн Г., Чойке П., Макклеллан М.В., Венг Ю., Дуан Д-СР, Дин М., Главац Д., Ричардс Ф.М., Кросси П.А., Фергюсон-Смит М.А., Ле Паслье Д., Чумаков И., Коэн Д. , Чинаулт А.Г., Махер Э.Р., Линехан В.М., Збар Б., Лерман М.И.1993 Наука 260 : 1317–1320
Линдал Т . 1993 Nature 362 : 709–715
Мандель Дж.С., Маклафлин Дж.К., Шлехофер Б., Меллемгаард А., Хелмерт У., Линдблад П., МакКреди М., Адами Х.О. 1995 Межд. Дж. Рак 61 : 601–605
Маккреди М., Поммер В., Маклафлин Дж.К., Стюарт Дж.Х., Линдблад П., Мандель Дж.С., Меллемгаард А., Шлехофер Б., Нива С. . 1995 Межд. Дж. Рак 60 : 345–349
Маклафлин Дж.К., Линдблад П., Меллемгаард А., МакКреди М., Мандель Дж.С., Шлехофер Б., Поммер В., Адами Х.О.1995 Межд. J. Рак 60 : 194–198
McLaughlin JK, Lipworth L . 2000 Семин. Oncol 27 : 115–123
Мейер А. Дж., Эрнандес А., Флорл А.Р., Энцманн Дж., Герхарц К.Д., Шульц В.А., Вернет П., Акерманн Р. . 2000 Межд. J. Рак 87 : 650–653
Дж., Эрнандес А., Флорл А.Р., Энцманн Дж., Герхарц К.Д., Шульц В.А., Вернет П., Акерманн Р. . 2000 Межд. J. Рак 87 : 650–653
Пауза А., Ли С., Уоррелл Р.А., Чен Д.Ю., Берджесс В.Х., Линехан В.М., Клауснер Р.Д. 1997 Проц. Натл. акад. науч. США 94 : 2156–2161
Проуз А.Х., Вебстер А.Р., Ричардс Ф.М., Ричард С., Ольшванг С., Реше Ф., Аффара Н.А., Махер Э.Р.1997 утра. Дж. Хам. Жене. 60 : 765–771
Schoenfeld AR, Davidowitz EJ, Burk RD . 2000a Proc. Натл. акад. науч. США 97 : 8507–8512
Шенфельд А.Р., Пэррис Т., Эйзенбергер А., Давидовиц Э.Дж., Де Леон М., Таласазан Ф., Девараджан П., Берк Р.Д. 2000b Oncogene 19 : 5851–5857
Schraml P, Zhaou M, Richter J, Bruning T, Pommer M, Sauter G, Mihatsch MJ, Moch H .1999 Верх. Дтч. Гэс. Патол. 83 : 218–224
Секидо Ю., Бадер С.А., Латиф Д.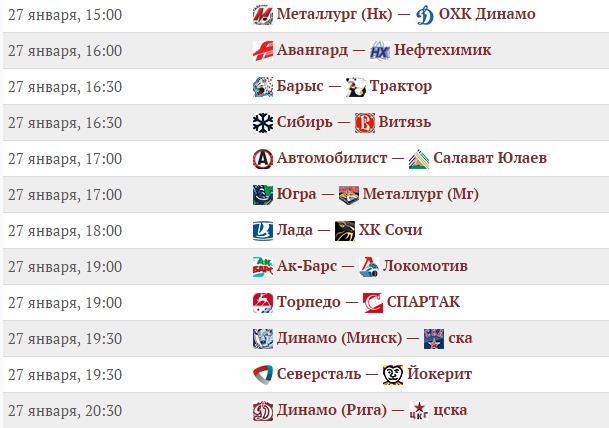 Р., Гнарра А.Ф., Газдар В.М., Линехан Б., Збар М.И., Лерман Д.Д., Минна Д.Д. 1994 Онкоген 9 : 1599–1604
Р., Гнарра А.Ф., Газдар В.М., Линехан Б., Збар М.И., Лерман Д.Д., Минна Д.Д. 1994 Онкоген 9 : 1599–1604
Шиао Ю.Х., Райс Дж.М., Андерсон Л.М., Диван Б.А., Hard GC. 1998 J. Natl. Рак инст. 90 : 1720–1723
Симейстер Г., Вейндель К., Морс К., Барлеон Б., Мартини-Барон Г., Марме Д. . 1996 Рак Рез. 56 : 2299–2301
Стеббинс К.Е., Каелин младший В.Г., Павлетич Н.П. 1999 Science 284 : 455–461
Thoenes W, Rumpelt HJ, Storkel S . 1990а Клин. Wochenschr. 68 : 1102–1111
Тонес В., Сторкель С., Румпелт Х.Дж., Молл Р. . 1990b Евро. Урол. 18 : 6–9
Ян К., Линдблад П., Эгевад Л., Хемминки К. . 1999 Рак Летт. 141 : 1–8
Ёсида М., Ашида С., Кондо К., Кобаяши К., Канно Х., Шинохара Н., Шитара Н., Кишида Т., Каваками С., Баба М., Ямамото И., Хосака М., Шуин Т. , Яо М. 2000 Япония. Дж. Рак Рез. 91 : 204–212
, Яо М. 2000 Япония. Дж. Рак Рез. 91 : 204–212
Zhuang Z, Gnarra JR, Dudley CF, Zbar B, Linehan WM, Lubensky IA. 1996 Мод. Патол. 9 : 838–842
Дефицит VHL вызывает энхансерную активацию онкогенов при светлоклеточной почечно-клеточной карциноме в 2012 г. по всему миру (1).Поскольку большинство зПКР являются радиохимиорезистентными, у пациентов с метастатическим зПКР показатель общей пятилетней выживаемости составляет 8% (2). Хотя таргетная терапия, ингибирующая ангиогенез и пути mTOR, может привести к начальному контролю над опухолью, у большинства пациентов резистентность развивается менее чем через год (3, 4). Таким образом, необходимо лучшее понимание молекулярных зависимостей и уязвимостей ccRCC для разработки новых терапевтических стратегий для пациентов, которым не удается стандартное лечение.
Потеря опухолевого супрессора фон Хиппеля-Линдау ( VHL ) является определяющим признаком скПКР (5, 6).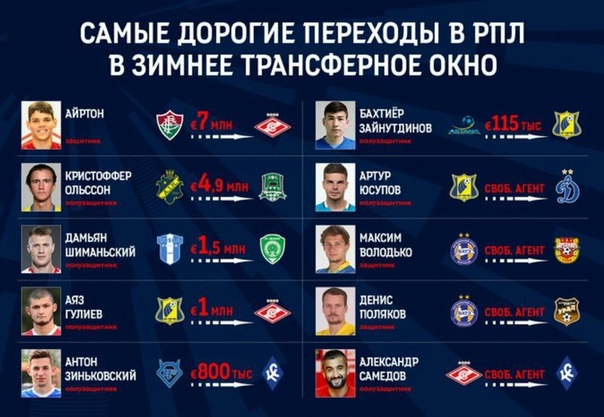 В сочетании с инактивацией дополнительных опухолевых супрессоров ( Pbrm1, Bap1, Trp53, Rb1 и/или Cdkn2a ) и/или активацией онкогенов ( Myc ) потеря Vhl вызывает спонтанное образование ccRCC в моделях мышей ( 7–11). VHL кодирует убиквитинлигазу E3 (12, 13), которая нацелена на деградацию HIF1α ( HIF1A ) и HIF2α ( EPAS1 ) (14, 15). Потеря VHL при скПКР приводит к конститутивной активации HIF1/2α и последующей трансактивации (16, 17) нижестоящих генов, регулирующих ангиогенез, гликолиз (18), липогенез (19, 20), клеточный цикл (21) и антиапоптоз (22). ).
В сочетании с инактивацией дополнительных опухолевых супрессоров ( Pbrm1, Bap1, Trp53, Rb1 и/или Cdkn2a ) и/или активацией онкогенов ( Myc ) потеря Vhl вызывает спонтанное образование ccRCC в моделях мышей ( 7–11). VHL кодирует убиквитинлигазу E3 (12, 13), которая нацелена на деградацию HIF1α ( HIF1A ) и HIF2α ( EPAS1 ) (14, 15). Потеря VHL при скПКР приводит к конститутивной активации HIF1/2α и последующей трансактивации (16, 17) нижестоящих генов, регулирующих ангиогенез, гликолиз (18), липогенез (19, 20), клеточный цикл (21) и антиапоптоз (22). ).
Большинство сообщений, изучающих активацию транскрипции VHL/HIF, сосредоточены на промоторах, связанных с HIF (23-28). Однако недавние данные свидетельствуют о новой роли дистальных энхансерных элементов в контроле транскрипции VHL/HIF (29, 30). Например, дистальные энхансеры, связанные с HIF2α, активируют протоонкогены MYC (31) и CCND1 (21) и совпадают с аллелями генетического риска ccRCC.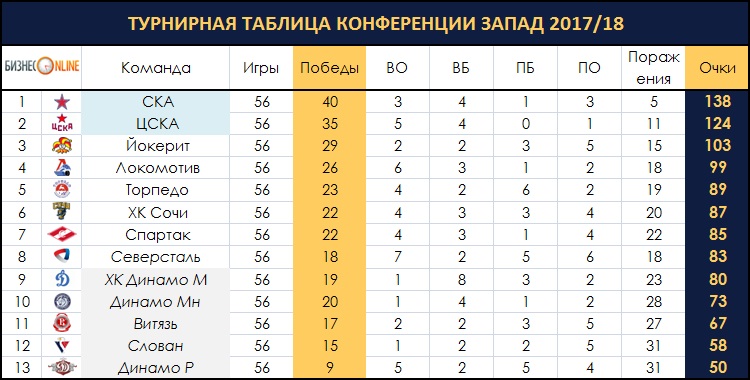 Тем не менее, такие исследования сосредоточены на индивидуальных энхансерах, и большинство дистальных элементов в ccRCC остаются в значительной степени неизученными.
Тем не менее, такие исследования сосредоточены на индивидуальных энхансерах, и большинство дистальных элементов в ccRCC остаются в значительной степени неизученными.
Очерчивание глобального регуляторного ландшафта ccRCC cis может также выявить главные регуляторы, участвующие в тканеспецифических патологических процессах. По сравнению с промоторами, которые в значительной степени инвариантны к типу клеток, дистальные энхансеры интегрируют множественные сигналы, зависящие от клона и контекста, удовлетворяя специализированные потребности различных типов клеток и заболеваний (32, 33). При раке такие главные регуляторы часто располагаются рядом с «суперэнхансерами» или «усилителями растяжения», отмеченными длинными участками сигналов h4K27ac (34, 35).Например, специфические для подтипа геномные изменения, такие как EGFRvIII при глиобластоме (36) и EWS-FLI при саркоме Юинга (37), индуцируют энхансеры de novo , вызывая реактивацию основных регуляторов развития, необходимых для самообновления и клонирования. спецификация (36). Хотя было показано, что инактивация VHL модулирует уровни белков различных модификаторов гистонов (например, KDM5C/JARID1C, ссылка 38; HDAC1, ссылка 39; JMJD1A, ссылка 40; JMJD2B, ссылка 40; JMJD2C, ссылка41), влияние этих белковых изменений на специфические эпигеномные локусы остается неясным. Кроме того, предыдущие исследования по профилированию модификаций гистонов при скПКР также были ограничены небольшими размерами выборки (2 случая; ссылка 42), зависимостью от систем in vitro , а также отсутствием интерактомных данных дальнего действия и тестирования функциональных энхансеров для точного определения родственные энхансерные мишени.
спецификация (36). Хотя было показано, что инактивация VHL модулирует уровни белков различных модификаторов гистонов (например, KDM5C/JARID1C, ссылка 38; HDAC1, ссылка 39; JMJD1A, ссылка 40; JMJD2B, ссылка 40; JMJD2C, ссылка41), влияние этих белковых изменений на специфические эпигеномные локусы остается неясным. Кроме того, предыдущие исследования по профилированию модификаций гистонов при скПКР также были ограничены небольшими размерами выборки (2 случая; ссылка 42), зависимостью от систем in vitro , а также отсутствием интерактомных данных дальнего действия и тестирования функциональных энхансеров для точного определения родственные энхансерные мишени.
В этом исследовании мы создали наиболее полную коллекцию профилей гистонов зПКР на сегодняшний день, аннотируя точное геномное расположение измененных промоторов, энхансеров и суперэнхансеров при зПКР.Используя изогенные клеточные линии с диким типом VHL или без него, мы дополнительно продемонстрировали, что помимо его четко определенной роли в восприятии кислорода, VHL также защищает ландшафт хроматина; его потеря индуцирует рост опухолеспецифических энхансеров вокруг характерных генов ccRCC, таких как ангиогенные и метаболические мишени, посредством стабилизации гетеродимеров HIF2α/HIF1β ( ARNT ) и рекрутирования гистон-ацетилтрансферазы p300 ( EP300 ).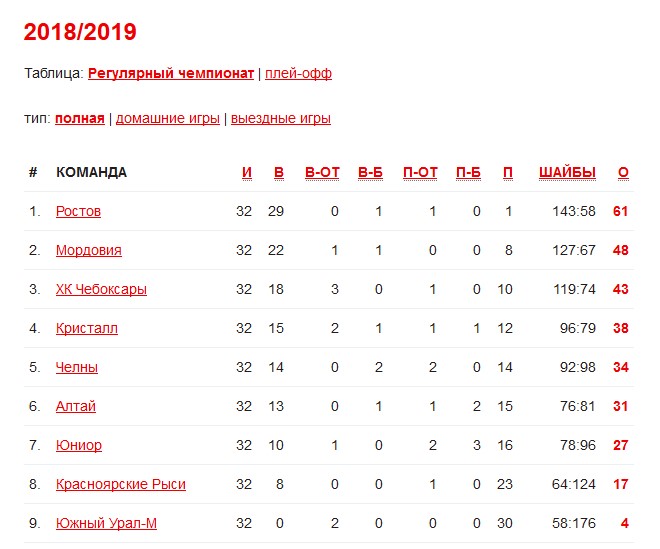 Одной важной мишенью эпигенетической активации является ZNF395 , главный регулятор онкогенеза ccRCC.Взятые в совокупности, наши результаты раскрывают эпигенетическую структуру, с помощью которой основная специфическая для ccRCC драйверная мутация, VHL , индуцирует энхансеров de novo , способствуя онкогенной транскрипции.
Одной важной мишенью эпигенетической активации является ZNF395 , главный регулятор онкогенеза ccRCC.Взятые в совокупности, наши результаты раскрывают эпигенетическую структуру, с помощью которой основная специфическая для ccRCC драйверная мутация, VHL , индуцирует энхансеров de novo , способствуя онкогенной транскрипции.
Результаты
Цис -Регуляторные ландшафты в опухолях зПКР являются аберрантными (3 балла: h4K27ac, h4K4me3 и h4K4me1) в 10 парах первичная опухоль/нормальная, 5 согласованных с пациентом опухолевых клеточных линиях, 2 коммерчески доступных линиях ccRCC (786-O и A-498) и 2 нормальных клеточных линиях почек (HK2 и PCS-400; см. дополнительную таблицу S1 для получения клинической информации о пациенте).Из исходных 87 образцов 79 образцов прошли фильтры контроля качества предварительного секвенирования и были подвергнуты обработке ChIP-seq и последующему анализу. В общей сложности мы сгенерировали 2 363 904 778 однозначно сопоставленных прочтений (см. Дополнительную таблицу S2 для статистики секвенирования). В среднем 89% пиков h4K27ac, 98% пиков h4K4me3 и 76% пиков h4K4me1, полученных в наших нормальных тканях почек, перекрывались с пиками из тканей почек взрослых в наборе данных Epigenomics Roadmap (дополнительный рисунок S1A). Среди 10 первичных ccRCC 9 содержали мутацию VHL , обнаруженную с помощью целевого секвенирования и подтвержденную секвенированием по Сэнгеру (дополнительная таблица S3).Клеточные линии 786-O и A-498 также содержат усеченные мутации VHL (дополнительная таблица S3). Мутации VHL сочетались с соматическими мутациями других модификаторов хроматина, обычно обнаруживаемых при скПКР, включая PBRM1 (7/10), SETD2 (1/10), KDM5A (1/10), KDM5C. (1/10), ARID1A (1/10) и KMT2C (1/10).
Дополнительную таблицу S2 для статистики секвенирования). В среднем 89% пиков h4K27ac, 98% пиков h4K4me3 и 76% пиков h4K4me1, полученных в наших нормальных тканях почек, перекрывались с пиками из тканей почек взрослых в наборе данных Epigenomics Roadmap (дополнительный рисунок S1A). Среди 10 первичных ccRCC 9 содержали мутацию VHL , обнаруженную с помощью целевого секвенирования и подтвержденную секвенированием по Сэнгеру (дополнительная таблица S3).Клеточные линии 786-O и A-498 также содержат усеченные мутации VHL (дополнительная таблица S3). Мутации VHL сочетались с соматическими мутациями других модификаторов хроматина, обычно обнаруживаемых при скПКР, включая PBRM1 (7/10), SETD2 (1/10), KDM5A (1/10), KDM5C. (1/10), ARID1A (1/10) и KMT2C (1/10). Специфические модификации гистонов могут различать различные категории функциональных регуляторных элементов — h4K4me3 обычно ассоциируется с промоторами, h4K4me1 с энхансерами и h4K27ac с активными элементами (33, 43). Интегрируя сигналы от трех гистоновых меток и аннотированных сайтов начала транскрипции (TSS) GENCODE v19, мы определили активные промоторы как области TSS h4K27ac + / h4K4me3 + / ± 2,0 т.п.н. области не перекрываются с промоторами. Сосредоточившись на эпигеномных событиях, характерных для соматических раковых клеток, мы получили клеточные линии пяти первичных опухолей и, в сочетании с коммерческими линиями, исключили пики, не обнаруженные ни в одной из клеточных линий, чтобы уменьшить смешанные эффекты стромальных клеток.В среднем мы наблюдали 80% перекрытие пиков ChIP-seq между первичными опухолями и соответствующими линиями (дополнительная рис. S1B). Используя эти критерии, мы идентифицировали 17 497 предполагаемых промоторов и 66 448 предполагаемых энхансеров (рис. 1А), что сопоставимо с предыдущими исследованиями других типов опухолей (43–45). Количество определенных промоторов и энхансеров достигло насыщения после 4 и 16 образцов, соответственно, что позволяет предположить, что размер выборки 20 (10 пар опухоль/нормальный) является достаточно мощным, чтобы обнаружить большинство цис -регуляторных элементов в ccRCC (дополнительная рис.
Интегрируя сигналы от трех гистоновых меток и аннотированных сайтов начала транскрипции (TSS) GENCODE v19, мы определили активные промоторы как области TSS h4K27ac + / h4K4me3 + / ± 2,0 т.п.н. области не перекрываются с промоторами. Сосредоточившись на эпигеномных событиях, характерных для соматических раковых клеток, мы получили клеточные линии пяти первичных опухолей и, в сочетании с коммерческими линиями, исключили пики, не обнаруженные ни в одной из клеточных линий, чтобы уменьшить смешанные эффекты стромальных клеток.В среднем мы наблюдали 80% перекрытие пиков ChIP-seq между первичными опухолями и соответствующими линиями (дополнительная рис. S1B). Используя эти критерии, мы идентифицировали 17 497 предполагаемых промоторов и 66 448 предполагаемых энхансеров (рис. 1А), что сопоставимо с предыдущими исследованиями других типов опухолей (43–45). Количество определенных промоторов и энхансеров достигло насыщения после 4 и 16 образцов, соответственно, что позволяет предположить, что размер выборки 20 (10 пар опухоль/нормальный) является достаточно мощным, чтобы обнаружить большинство цис -регуляторных элементов в ccRCC (дополнительная рис.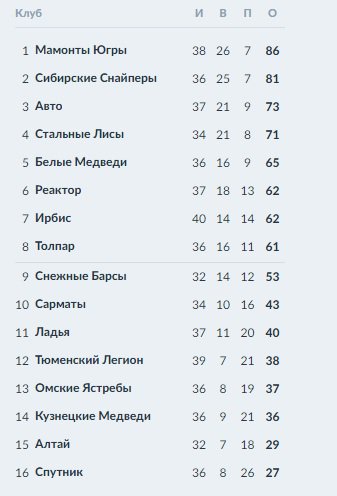 .S1C и S1D). Анализ основных компонентов (PCA) с использованием первых двух компонентов глобальной интенсивности h4K27ac на промоторах или энхансерах (составляющих 83% и 64% общей дисперсии соответственно; дополнительные рис. S1E и S1F) успешно разделил нормальные и опухолевые образцы, что указывает на то, что геном- широкие первазивные изменения в цис -регуляторных элементах являются отличительной особенностью ccRCC (Fig. 1B).
.S1C и S1D). Анализ основных компонентов (PCA) с использованием первых двух компонентов глобальной интенсивности h4K27ac на промоторах или энхансерах (составляющих 83% и 64% общей дисперсии соответственно; дополнительные рис. S1E и S1F) успешно разделил нормальные и опухолевые образцы, что указывает на то, что геном- широкие первазивные изменения в цис -регуляторных элементах являются отличительной особенностью ccRCC (Fig. 1B).
Опухоли ccRCC с дефицитом VHL демонстрируют аберрантный цис -регуляторный ландшафт. A, Предполагаемые активные промоторы определяются совместным присутствием h4K4me3, h4K27ac и близостью к TSS в пределах 2 т.п.о. Предполагаемые активные энхансеры определяются наличием h4K4me1, h4K27ac и эксклюзивностью промоторов. Анализ PCA B, с использованием всех 17 497 промоторов и 66 448 энхансеров классифицирует норму и опухоль на отдельные кластеры. Идентификаторы пациентов: 1-12364284; 2-17621953; 3-20431713; 4-40
2; 5-57398667; 6-70528835; 7-74575859; 8-77972083; 9-86049102; 10-75416923. C, Тепловые карты показывают уровни h4K27ac измененных промоторов и энхансеров в парной ткани пациента (пациент 40
C, Тепловые карты показывают уровни h4K27ac измененных промоторов и энхансеров в парной ткани пациента (пациент 40
2; желтый высокий, черный низкий). D, Уровни h4K27ac, доступность хроматина (FAIRE-seq), метилирование ДНК полученных промоторов и энхансеров и экспрессия гена ближайшей днРНК ccRCC сравнивают в нормальных и опухолевых тканях. ***, P < 0,001, двусторонний тест t . E, Показаны следы секвенирования ChIP-seq гистонов (h4K27ac, h4K4me1 и h4K4me3) и секвенирования РНК (RNA-seq) в локусе CCND1 в паре опухоль-нормальный пациент 40
2. Профили ChIP-seq гистонов нормальной ткани почки взрослого человека из дорожной карты Epigenome отображаются над нормальной тканью, созданной с помощью Nano-ChIP-seq, для сравнения.Клеточная линия была получена из опухолевой ткани, и ее гистоновый профиль показан под соответствующей первичной тканью. Известно, что этот энхансер взаимодействует с промотором CCND1 из предыдущего исследования (21) и расположен близко к SNP rs7105934 восприимчивости к RCC (49).
Мы провели дифференциальный анализ для выявления измененных промоторов и энхансеров. Чтобы определить приобретенные или потерянные области, мы применили кратную разницу h4K27ac RPKM ≥ 2, абсолютную разницу ≥ 0,5 и для большей строгости отсутствие изменений в обратном направлении в оставшихся парах опухоль/нормальный (см.S1G для распределения измененных элементов по количеству пациентов). На пороге ≥5/10 пациентов 80% измененных областей достигли статистической значимости ( q -значение <0,1, парный тест t , с поправкой Бенджамини-Хохберга; дополнительная рис. S1H), и при этом же пороговое значение, увеличение доли образцов, отвечающих статистической значимости, достигло седловой точки (дополнительный рис. S1I). Применяя эти критерии, мы получили высоконадежный и полный набор из 4719 приобретенных промоторов, 592 потерянных промоторов, 4906 полученных энхансеров и 5654 потерянных энхансеров (рис.1А и С; Дополнительная таблица S4). Репрезентативные регионы представлены на дополнительном рисунке S2.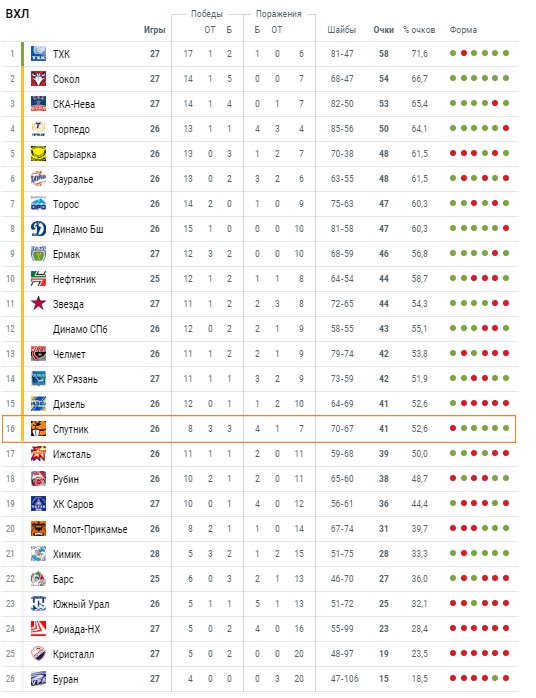
Подтверждая надежность этих данных, полученные промоторы и энхансеры демонстрировали повышенную доступность хроматина, измеренную более высокими сигналами FAIRE-seq (46) в опухолевых тканях, чем в нормальных тканях, соответственно ( P < 0,0001), а также снижали метилирование ДНК на основе данных из Атласа генома рака (TCGA; ссылка 47), что согласуется с взаимными отношениями между активными регуляторными областями и метилированием ДНК (рис.1Д). Интересно, что мы также отметили повышенную экспрессию длинных некодирующих РНК (48), прилегающих к приобретенным промоторам и энхансерам, в опухолевых тканях по сравнению с нормальными тканями ( P < 0,0001 соответственно). Наконец, мы подтвердили, что многие из наших цис -регуляторных элементов включали области, ранее вовлеченные в скПКР; например, мы наблюдали усиление сигналов h4K27ac и обогащение h4K4me1 в дистальном энхансере CCND1 , перекрывающемся с локусом восприимчивости к RCC (rs7105934; refs. 21 и 49; Рис. 1Е). Наша способность заново открывать этот важный усилитель в нашем беспристрастном профилировании поддерживает надежность наших данных.
21 и 49; Рис. 1Е). Наша способность заново открывать этот важный усилитель в нашем беспристрастном профилировании поддерживает надежность наших данных.
Опухолеспецифические энхансеры связаны с признаками скПКР
Для идентификации генов, модулируемых опухолеспецифическими регуляторными элементами, мы распределили энхансеры, используя три подхода. В первом подходе использовались предопределенные правила линейной близости, включающие набор высоконадежных генов (алгоритм GREAT; ссылка 50; дополнительная таблица S5). Анализ пути MSigDB с использованием генов, назначенных GREAT, показал, что полученные энхансеры демонстрируют очень значимую специфичную для ПКР сигнатуру по сравнению с полученными промоторами (значение энхансера q = 3.2 × 10 −26 ; промотор q — значение = 1,5 × 10 -1 , биномиальный FDR; Рис. 2А). Хотя полученные промоторы были вовлечены в общие процессы рака (например, клеточный цикл, транскрипцию и метаболизм РНК; полный список путей промоторов см. в дополнительной таблице S6), полученные энхансеры были обогащены специфическими для заболевания особенностями ccRCC, включая сетевую активность HIF1α. , проангиогенные пути (активация тромбоцитов и передача сигналов PDGFRβ) и SLC-опосредованный трансмембранный транспорт (фиг.2А; Дополнительная таблица S7 для полного списка путей энхансера). Примечательно, что сетевая активность HIF1α неизменно фигурирует в качестве одного из пяти основных путей, даже с нарушениями пороговых значений пациентов, используемых для определения полученных энхансеров (≥3–8 пациентов; дополнительная таблица S8).
в дополнительной таблице S6), полученные энхансеры были обогащены специфическими для заболевания особенностями ccRCC, включая сетевую активность HIF1α. , проангиогенные пути (активация тромбоцитов и передача сигналов PDGFRβ) и SLC-опосредованный трансмембранный транспорт (фиг.2А; Дополнительная таблица S7 для полного списка путей энхансера). Примечательно, что сетевая активность HIF1α неизменно фигурирует в качестве одного из пяти основных путей, даже с нарушениями пороговых значений пациентов, используемых для определения полученных энхансеров (≥3–8 пациентов; дополнительная таблица S8).
Enhancer является сигнатурой ccRCC. A, Обогащенные пути, связанные с полученными промоторами и энхансерами, выявленные с помощью GREAT (ранжированы по биномиальному значению FDR q ). Красные столбцы относятся к специфическим для скПКР путям. B, Энхансеры De novo приобретаются в опухолевой ткани ccRCC выше VEGFA .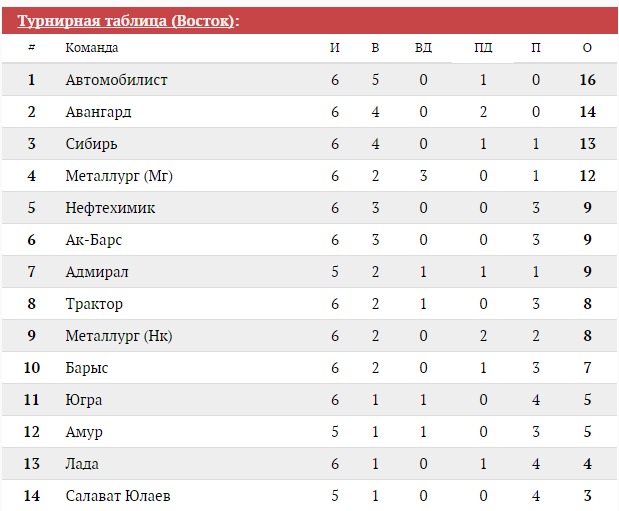 Capture-C подтвердил взаимодействие этого энхансера VEGFA (E) с его промотором (P) в клетках 786-O. Дуги представляют значительные взаимодействия, обнаруженные с помощью r3Cseq (q <0,05). Сигналы h4K27ac этого энхансера после вычитания входных данных сильно коррелируют с экспрессией гена VEGFA (корреляция Спирмена). C, Подобно ( B ), энхансер опухоли de novo взаимодействует с промотором SLC2A1/GLUT1 .
Capture-C подтвердил взаимодействие этого энхансера VEGFA (E) с его промотором (P) в клетках 786-O. Дуги представляют значительные взаимодействия, обнаруженные с помощью r3Cseq (q <0,05). Сигналы h4K27ac этого энхансера после вычитания входных данных сильно коррелируют с экспрессией гена VEGFA (корреляция Спирмена). C, Подобно ( B ), энхансер опухоли de novo взаимодействует с промотором SLC2A1/GLUT1 .
Отдельные гены, связанные с приобретенными энхансерами, включали хорошо известные гипоксические мишени ( VEGFA , рис. 2B; CXCR4 ) и метаболические гены, участвующие в гликолизе, потреблении глутамина и накоплении липидов ( GLUT1/SLC2A1 , рис. 2C). ; HK2, PFKFB3, PLIN2 , дополнительный рисунок S3A) и SLC38A1 (дополнительный рисунок S3B; ссылка 51). Присутствие энхансеров вокруг метаболических ферментов и транспортеров в значительной степени согласуется с метаболическим контекстом ccRCC, который включает повышенный гликолиз и глутаминолиз (19, 52-55). Действительно, анализ генной онтологии (GO) полученных энхансеров сильно отразил характерные метаболические изменения, связанные с ccRCC, включая активность трансмембранного транспортера монокарбоновой кислоты (биномиальное значение FDR q = 1,6 × 10 -10 ; дополнительная рис. S3C).
Действительно, анализ генной онтологии (GO) полученных энхансеров сильно отразил характерные метаболические изменения, связанные с ccRCC, включая активность трансмембранного транспортера монокарбоновой кислоты (биномиальное значение FDR q = 1,6 × 10 -10 ; дополнительная рис. S3C).
Мы также использовали второй метод назначения гена-энхансера, основанный на корреляции между сигналами h4K27ac и экспрессией генов в пределах одного и того же топологически ассоциированного домена (TAD; ссылка 34). Используя q -значение <0.05 на основе корреляции Спирмена мы назначили 2311 полученных энхансеров 2186 мишеням, кодирующим белок (дополнительная таблица S9). Обнадеживает, что сигналы h4K27ac многих полученных энхансеров сильно коррелируют с экспрессией генов их предполагаемых генов-мишеней. Например, уровни h4K27ac энхансера VEGFA показали высокую корреляцию с экспрессией гена VEGFA ( r = 0,83, корреляция Спирмена), тогда как сигналы h4K27ac энхансера SLC2A1 сильно коррелировали с экспрессией гена SLC2A1 9041 ( r = 0,83, корреляция Спирмена).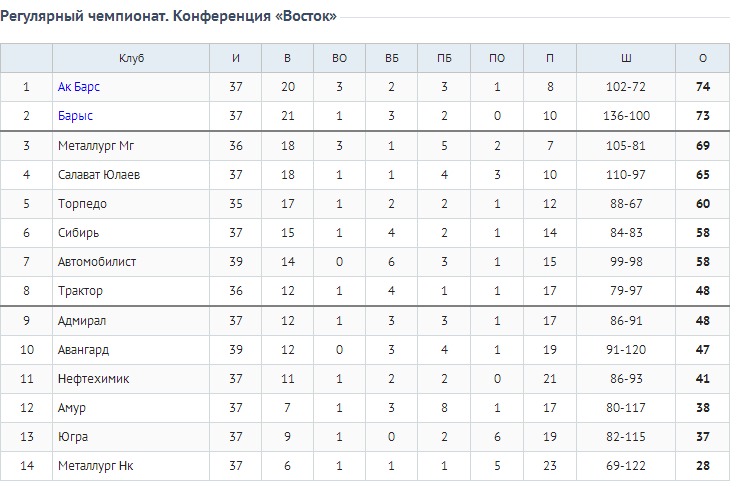 г = 0.72, корреляция Спирмена; рис. 2Б; Дополнительный рис. S3D). Подобно подходу GREAT, корреляционный подход TAD также выделял гипоксию (Krieg_Hypoxia_not_via_KDM3A, значение FDR q = 7 × 10 −120 ) и метаболизм (Chen_Metabolic_Syndrome_Network, значение FDR q = 2 × 410 94 94 ). ) как высокообогащенные пути (дополнительная таблица S10).
г = 0.72, корреляция Спирмена; рис. 2Б; Дополнительный рис. S3D). Подобно подходу GREAT, корреляционный подход TAD также выделял гипоксию (Krieg_Hypoxia_not_via_KDM3A, значение FDR q = 7 × 10 −120 ) и метаболизм (Chen_Metabolic_Syndrome_Network, значение FDR q = 2 × 410 94 94 ). ) как высокообогащенные пути (дополнительная таблица S10).
В-третьих, для независимой проверки подходов GREAT и TAD в конкретном контексте ccRCC мы экспериментально исследовали интерактом опухолеспецифических энхансеров ccRCC, выполнив анализы Capture-C (56).По сравнению с другими методами захвата хроматина Capture-C предлагает как высокое разрешение (до разрешения в один килобайт), так и высокую пропускную способность опроса определяемых пользователем областей (обычный рабочий диапазон 10–500 областей). Мы разработали зонды против подмножества из 56 полученных энхансеров и исследовали их взаимодействие с генами, кодирующими белок, в клетках 786-O. Каждая пара ген-энхансер, обнаруженная с помощью Capture-C, была дополнительно отфильтрована по корреляциям между экспрессией гена и уровнями h4K27ac (значение q < 0. 05). 56 полученных энхансеров были соединены с 36 генами, кодирующими белок (дополнительная таблица S11), из них 58% были предсказаны GREAT, а 80% — корреляциями генов в пределах TAD. Среднее расстояние взаимодействий, обнаруженных Capture-C, составляло 16 КБ, а 83% взаимодействий приходилось на окно размером 100 КБ (дополнительный рисунок S3E). В качестве наглядного примера Capture-C подтвердил взаимодействие между энхансером VEGFA и TSS VEGFA на расстоянии ~100 т.п.н. (рис. 2B), а также взаимодействие между энхансером SLC2A1 и его промотором (рис.2С). Взятые в совокупности, эти данные подчеркивают специфическую для заболевания природу энхансерных элементов (33) и важную роль нарушения функции энхансера в модулировании патологии ccRCC.
05). 56 полученных энхансеров были соединены с 36 генами, кодирующими белок (дополнительная таблица S11), из них 58% были предсказаны GREAT, а 80% — корреляциями генов в пределах TAD. Среднее расстояние взаимодействий, обнаруженных Capture-C, составляло 16 КБ, а 83% взаимодействий приходилось на окно размером 100 КБ (дополнительный рисунок S3E). В качестве наглядного примера Capture-C подтвердил взаимодействие между энхансером VEGFA и TSS VEGFA на расстоянии ~100 т.п.н. (рис. 2B), а также взаимодействие между энхансером SLC2A1 и его промотором (рис.2С). Взятые в совокупности, эти данные подчеркивают специфическую для заболевания природу энхансерных элементов (33) и важную роль нарушения функции энхансера в модулировании патологии ccRCC.
Опухолевые суперэнхансеры Идентификация
ZNF395 в качестве основного регулятора скПКР Важность энхансеров в скПКР привела нас к изучению ландшафта «суперэнхансеров» или «усилителей растяжения» — плотных кластеров энхансеров, расположенных рядом с основными регуляторами клеточного личность и болезнь (35, 57). Используя ROSE (35), мы определили 1451 суперэнхансер в когорте ccRCC, из которых 1157 были получены в опухолях, а 294 были потеряны в опухолях (дополнительная таблица S12).
Используя ROSE (35), мы определили 1451 суперэнхансер в когорте ccRCC, из которых 1157 были получены в опухолях, а 294 были потеряны в опухолях (дополнительная таблица S12).
Предполагаемые мишени высокоэффективных суперэнхансеров подтверждены хорошо известными онкогенами, включая MYC/PVT1, VEGFA и HIF2A (рис. 3A; дополнительные рис. S4A и S4B). Кроме того, мы обнаружили несколько менее известных генов, включая ERGIC1, ZNF395, SLC28A1 и SMPDL3A (рис. 3B). Эти гены были сильно сверхэкспрессированы в опухолях по сравнению с соответствующими им нормальными тканями (рис.3Б). Кроме того, они были уникальными для ccRCC и не были сверхэкспрессированы в папиллярных и хромофобных RCC, двух других различных подтипах ccRCC (рис. 3B). Например, ZNF395 продемонстрировал соотношение опухоль-норма ~7 при скПКР ( P = 1 × 10 -22 , парный тест t ), но имел небольшую гиперэкспрессию при папиллярном и хромофобном ПКР с соотношением опухоль-норма.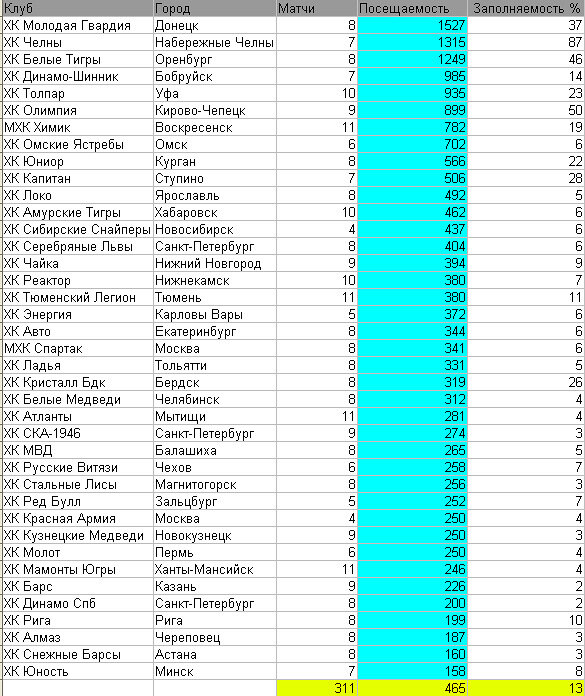 1,2 и 1,3 соответственно ( P = 0,02 в папиллярном и P = 0,06 в хромофобном, парный тест t ).
1,2 и 1,3 соответственно ( P = 0,02 в папиллярном и P = 0,06 в хромофобном, парный тест t ).
Опухолевые суперэнхансеры (SE) определяют ключевые факторы онкогенности. A, В общей сложности 1451 суперэнхансер идентифицирован ROSE и ранжирован по их дифференциальной интенсивности h4K27ac между нормальными и опухолевыми тканями. Перечислены гены, связанные с главными приобретенными и потерянными суперэнхансерами. B, Данные TCGA RNA-seq показывают, что гены, связанные с 10 основными полученными энхансерами, активируются в опухолях, тогда как гены, связанные с 10 основными потерянными энхансерами, подавляются.Эта опухолевая специфичность ограничивается скПКР, но не двумя другими подтипами ПКР, папиллярным и хромофобным. C, Экспрессия ZNF395 и SMPDL3A измерены на панели нормальных клеточных линий почек (черные) и клеточных линий ccRCC (красные) с помощью RT-qPCR. D, Пул миРНК против ZNF395 ингибирует образование колоний A-498 и 786-O, но не нормальных иммортализованных клеток почек HK2. Объединенная миРНК против SMPLD3A ингибирует образование колоний A-498, но не 786-O. E, h4K27ac ChIP-seq показывает активный суперэнхансер ZNF395 только в клетках ccRCC (A-498 и 786-O), но не в нормальных клетках почек (PCS-400, HK2). Данные секвенирования РНК F, TCGA показывают исключительную сверхэкспрессию ZNF395 среди 12 типов рака. G–I, ZNF395 ингибирование двумя клонами кшРНК снижает образование колоний ( G ) и пролиферацию in vitro ( H ) и увеличивает апоптоз, измеряемый расщеплением субстрата каспазы 3/7 (
Объединенная миРНК против SMPLD3A ингибирует образование колоний A-498, но не 786-O. E, h4K27ac ChIP-seq показывает активный суперэнхансер ZNF395 только в клетках ccRCC (A-498 и 786-O), но не в нормальных клетках почек (PCS-400, HK2). Данные секвенирования РНК F, TCGA показывают исключительную сверхэкспрессию ZNF395 среди 12 типов рака. G–I, ZNF395 ингибирование двумя клонами кшРНК снижает образование колоний ( G ) и пролиферацию in vitro ( H ) и увеличивает апоптоз, измеряемый расщеплением субстрата каспазы 3/7 (
4 I 9) .*,
P < 0,05, двусторонний тест t . J, ZNF395 ингибирование кшРНК приводит к полной элиминации опухолей A-498 in vivo и замедлению роста опухоли 786-O. NC: n = 7; ш ЗНФ395 -1: н = 7; sh ZNF395 -2: n = 6. Напротив, гены, связанные с утратой суперэнхансеров, были рекуррентно подавлены при скПКР и включали EFHD1, EHF, MAL, GCOM1 и HOXB9 (рис. 3Б). В отличие от линейно-специфической природы опухолевых суперэнхансеров, гены, связанные с потерянными суперэнхансерами, были общими для зПКР и папиллярного ПКР, что указывает на более универсальную функцию генов-супрессоров опухолей. Например, EHF/ESE2 , супрессор опухоли, ранее обнаруженный при раке предстательной железы (58, 59), демонстрировал сниженную экспрессию во всех трех подтипах ПКР (опухоль ccRCC/норма = 0,05, P = 3 × 10 -15 ; папиллярная опухоль/норма = 0,1, P 2 × 10 -6 ; хромофобная опухоль/норма = 0.1, P = 2 × 10 −6 ).
3Б). В отличие от линейно-специфической природы опухолевых суперэнхансеров, гены, связанные с потерянными суперэнхансерами, были общими для зПКР и папиллярного ПКР, что указывает на более универсальную функцию генов-супрессоров опухолей. Например, EHF/ESE2 , супрессор опухоли, ранее обнаруженный при раке предстательной железы (58, 59), демонстрировал сниженную экспрессию во всех трех подтипах ПКР (опухоль ccRCC/норма = 0,05, P = 3 × 10 -15 ; папиллярная опухоль/норма = 0,1, P 2 × 10 -6 ; хромофобная опухоль/норма = 0.1, P = 2 × 10 −6 ).
Поскольку текущие терапевтические мишени при раке почки ограничены путями ангиогенеза и mTOR (3), мы стремились изучить эти менее изученные гены, обнаруженные с помощью профилирования суперэнхансеров. Мы выбрали ZNF395 и SMPDL3A из-за их дифференциальной опухолевой экспрессии (соотношение опухоль-нормаль 6–7; рис. 3B) и высокой распространенности (среднее RPKM ZNF395 ~ 112; среднее RPKM SMPDL3A ~ 58). Несмотря на то, что ZNF395 ранее был идентифицирован как потенциальный биомаркер скПКР (60), его функциональная роль в злокачественных новообразованиях скПКР остается неизученной. SMPDL3A имеет 31% аминокислотную идентичность с кислой сфингомиелиназой SMPD1 и является мишенью основного регулятора метаболизма холестерина, Х-рецепторов печени (LXR; ссылка 61).
Несмотря на то, что ZNF395 ранее был идентифицирован как потенциальный биомаркер скПКР (60), его функциональная роль в злокачественных новообразованиях скПКР остается неизученной. SMPDL3A имеет 31% аминокислотную идентичность с кислой сфингомиелиназой SMPD1 и является мишенью основного регулятора метаболизма холестерина, Х-рецепторов печени (LXR; ссылка 61).
Количественная ПЦР (рис. 3C) и иммуноблоттинг (дополнительная рис. S4C) подтвердили, что клетки ccRCC A-498 и 786-O демонстрируют высокую экспрессию ZNF395 и SMPDL3A , тогда как нормальные клетки проксимальных канальцев почек, PCS-400 и HK2, проявляли низкую экспрессию обоих генов.Опосредованный киРНК нокдаун SMPDL3A оказывал зависящее от клеточной линии влияние на образование колоний, ингибируя рост клеток A-498, но не оказывая заметного влияния на клетки 786-O (фиг. 3D). С другой стороны, ZNF395 постоянно ингибировал образование колоний как в клетках 786-O, так и в клетках A-498, но оказывал минимальное влияние на нормальные клетки почек (рис. 3D; дополнительная рис. S4D). В соответствии с этим фенотипическим наблюдением суперэнхансер ZNF395 был активен только в клетках ccRCC (786-O и A-498), но молчал в нормальных клетках почек (HK2 и PCS-400; рис.3Е). Кроме того, среди 12 типов рака, профилированных с помощью TCGA, ZNF395 был исключительно сверхэкспрессирован в опухолях ccRCC, что согласуется с предполагаемой клонально-специфической природой суперэнхансеров (рис. 3F).
3D; дополнительная рис. S4D). В соответствии с этим фенотипическим наблюдением суперэнхансер ZNF395 был активен только в клетках ccRCC (786-O и A-498), но молчал в нормальных клетках почек (HK2 и PCS-400; рис.3Е). Кроме того, среди 12 типов рака, профилированных с помощью TCGA, ZNF395 был исключительно сверхэкспрессирован в опухолях ccRCC, что согласуется с предполагаемой клонально-специфической природой суперэнхансеров (рис. 3F).
На сегодняшний день ни одно исследование функционально не тестировало туморогенное требование ZNF395 при ccRCC или любом другом типе рака. Мы подтвердили эффект стимулирования опухоли ZNF395 с использованием отдельных клонов кшРНК (дополнительные рисунки S4E и S4F). Два независимых клона кшРНК ZNF395 резко снижали образование колоний in vitro (рис.3G) и жизнеспособность клеток (фиг. 3H) как в клетках A-498, так и в клетках 786-O. Нокдаун ZNF395 также приводил к усилению апоптоза, измеренному расщеплением субстратов каспазы 3/7 (рис. 3I) и окрашиванием аннексина V (дополнительная рис. S4G). Исследования формирования опухоли in vivo в моделях ксенотрансплантата мыши выявили заметное подавление опухоли за счет истощения ZNF395 (фиг. 3J). Нокдаун ZNF395 привел к элиминации опухолей A-498 до 74-го дня, когда опухоли в контрольной группе начали превышать пределы размера, установленные протоколами для животных в учреждениях.Точно так же истощение ZNF395 значительно замедляло in vivo рост опухоли клеток 786-O (фиг. 3J). В совокупности мы показали незаменимую роль ZNF395 в онкогенезе ccRCC.
3I) и окрашиванием аннексина V (дополнительная рис. S4G). Исследования формирования опухоли in vivo в моделях ксенотрансплантата мыши выявили заметное подавление опухоли за счет истощения ZNF395 (фиг. 3J). Нокдаун ZNF395 привел к элиминации опухолей A-498 до 74-го дня, когда опухоли в контрольной группе начали превышать пределы размера, установленные протоколами для животных в учреждениях.Точно так же истощение ZNF395 значительно замедляло in vivo рост опухоли клеток 786-O (фиг. 3J). В совокупности мы показали незаменимую роль ZNF395 в онкогенезе ccRCC.
Чтобы изучить степень, в которой эпигенетические изменения, наблюдаемые в первичных ccRCC (рис. 1), непосредственно обусловлены потерей VHL , мы исследовали изменения хроматина в изогенных клеточных линиях с и без него. Восстановление ВХЛ .В соответствии с более ранними функциональными исследованиями VHL (62–64), восстановление VHL в клетках 786-O, A-498 и 12364284 оказывало незначительное влияние на пролиферацию, образование колоний и апоптоз in vitro , но сильно задерживалось. рост опухоли in vivo (дополнительная рис. S5A – S5D), что свидетельствует о важности VHL в модуляции процессов, необходимых для in vivo онкогенеза, включая перекрестные помехи опухоль-строма, ангиогенез, взаимодействия клетки-матрицы или опухоль метаболизм.
рост опухоли in vivo (дополнительная рис. S5A – S5D), что свидетельствует о важности VHL в модуляции процессов, необходимых для in vivo онкогенеза, включая перекрестные помехи опухоль-строма, ангиогенез, взаимодействия клетки-матрицы или опухоль метаболизм.
Сосредоточив внимание на тех же областях, которые определены в первичных опухолях (4719 промоторов, 4906 энхансеров и 1157 суперэнхансеров; рис. 1A), мы исследовали VHL -управляемые изменения h4K27ac в четырех различных клеточных линиях (две коммерческие клеточные линии : 786-0 и А-498 и две клеточные линии, полученные от пациентов: 12364284 и 40
2). Последовательно во всех четырех клеточных линиях восстановление VHL вызывало более выраженные изменения на энхансерах и суперэнхансерах, чем на промоторах (рис.4А; Дополнительный рис. S6A – S6C). Например, в клетках 786-О после восстановления VHL 12% энхансеров (549 энхансеров) были значительно истощены по сравнению с 6,5% промоторов (321 промотер; рис. 4А). Мы подтвердили, что большая доля энхансеров была значительно изменена при восстановлении VHL , чем промоторы ( P < 2,2 × 10 -16 , тест пропорций), и даже более высокая доля была связана с приобретенными суперэнхансерами ( P < 2,2 × 10 −16 , проверка пропорций).
4А). Мы подтвердили, что большая доля энхансеров была значительно изменена при восстановлении VHL , чем промоторы ( P < 2,2 × 10 -16 , тест пропорций), и даже более высокая доля была связана с приобретенными суперэнхансерами ( P < 2,2 × 10 −16 , проверка пропорций).
Дефицит VHL ремоделирует энхансеры ccRCC. A, Логарифмическая кратность изменений сигналов h4K27ac ChIP-seq на полученных промоторах, энхансерах и суперэнхансерах, как определено в первичном наборе данных ccRCC после восстановления VHL в клетках 786-O. Красные точки представляют цис -регуляторные элементы со значительными изменениями ( P <0,05, отрицательный бином) уровней h4K27ac после восстановления VHL . Количество и процент измененных областей ( P < 0.05, отрицательный бином) показаны в правом верхнем и нижнем углах. EV, пустой вектор. B, Считывание охвата h4K27ac ChIP-seq на VHL -чувствительные энхансеры в VHL — мутантные клеточные линии ccRCC (красные) по сравнению с VHL — ccRCC дикого типа (серые) и нормальные клеточные линии почек (зеленые) ) и 31 другая линия раковых клеток (черные).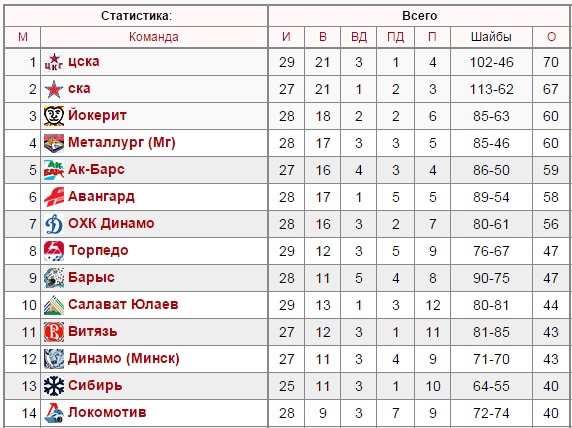 C, Изменения в экспрессии генов, связанных с VHL -чувствительными опухолевыми энхансерами, после восстановления VHL в клетках 786-O.*, P < 0,05, двусторонний тест t . D–G, Примеры потерянных VHL-чувствительных энхансеров/суперэнхансеров связаны с EGFR ( D ), CCND1 ( E ), ITGB3 (
C, Изменения в экспрессии генов, связанных с VHL -чувствительными опухолевыми энхансерами, после восстановления VHL в клетках 786-O.*, P < 0,05, двусторонний тест t . D–G, Примеры потерянных VHL-чувствительных энхансеров/суперэнхансеров связаны с EGFR ( D ), CCND1 ( E ), ITGB3 (
3 F
), G ) в ячейках 786-О. H, Логарифмическая кратность изменений сигналов h4K27ac (красный), h4K4me1 (синий) и h4K27me3 (коричневый) на полученных энхансерах, показывающая истощение h4K27ac после восстановления VHL в клетках 786-O. Хотя ожидалось, что полученные энхансеры будут демонстрировать только истощение после восстановления VHL , изменения уровней h4K27ac были двунаправленными (рис. 4А). Однако только полученные энхансеры с истощением h4K27ac были уникально активны в VHL -мутированных клеточных линиях ccRCC (786-O, A-498 и 12364284) по сравнению с VHL – клетками ccRCC дикого типа (86049102L), нормальными клетками почек. линии (PCS-400, HK2 и HKC-8) и 31 другая клеточная линия различных типов рака (рис. 4Б). Отсутствие сигналов h4K27ac в нормальных клеточных линиях почек свидетельствует о том, что тканевое происхождение является доминирующим источником высоких сигналов h4K27ac ChIP-seq, наблюдаемых в клеточных линиях ccRCC.С другой стороны, полученные энхансеры с обогащением h4K27ac после восстановления VHL показали высокую активность в отношении нескольких типов рака, что позволяет предположить, что эти энхансеры не являются уникальными для ccRCC (рис. 4B).
линии (PCS-400, HK2 и HKC-8) и 31 другая клеточная линия различных типов рака (рис. 4Б). Отсутствие сигналов h4K27ac в нормальных клеточных линиях почек свидетельствует о том, что тканевое происхождение является доминирующим источником высоких сигналов h4K27ac ChIP-seq, наблюдаемых в клеточных линиях ccRCC.С другой стороны, полученные энхансеры с обогащением h4K27ac после восстановления VHL показали высокую активность в отношении нескольких типов рака, что позволяет предположить, что эти энхансеры не являются уникальными для ccRCC (рис. 4B).
Кроме того, только полученные энхансеры, показывающие истощение h4K27ac после восстановления VHL , были в значительной степени связаны с сопутствующим снижением экспрессии генов их предполагаемых мишеней как в клетках 786-O, так и в клетках 12364284, тогда как энхансеры, полученные в первичных ccRCC и дополнительно обогащенные h4K27ac после Восстановление VHL не привело к значимой активизации гена на глобальном уровне (рис. 4С; Дополнительный рис. S6D). Эти результаты позволяют предположить, что первые энхансеры (истощение h4K27ac), вероятно, представляют собой эпигеномные изменения, специфичные для ccRCC и VHL , тогда как последние энхансеры (обогащение h4K27ac), вероятно, представляют общие компенсаторные механизмы в ответ на восстановление VHL . .
4С; Дополнительный рис. S6D). Эти результаты позволяют предположить, что первые энхансеры (истощение h4K27ac), вероятно, представляют собой эпигеномные изменения, специфичные для ccRCC и VHL , тогда как последние энхансеры (обогащение h4K27ac), вероятно, представляют общие компенсаторные механизмы в ответ на восстановление VHL . .
Комбинируя данные из нескольких линий, в общей сложности 1564 энхансера были истощены восстановлением VHL в ≥1 клеточной линии, что составляет почти треть (32%) всех полученных энхансеров, идентифицированных в первичных опухолях ccRCC (дополнительная таблица S13).Доля VHL -чувствительных энхансеров увеличивалась с уровнем рецидива у пациентов — только у 7,8% нерецидивно полученных энхансеров (1/10 пациентов) наблюдалось VHL -опосредованное истощение h4K27ac, тогда как 18% энхансеров повторно приобретались в 9 из 10 случаев. пациентов, и 20% энхансеров, полученных у 10 из 10 пациентов, показали истощение h4K27ac в клетках 786-O (дополнительный рисунок S7A, P = 0,0001, тест пропорций), что согласуется с высокой распространенностью мутаций VHL (9/10 пациентов) в нашем наборе открытий. Интересно, что неконтролируемая кластеризация с использованием 1564 VHL -чувствительных полученных энхансеров отделила одну VHL -опухоль дикого типа (ID 75416923) от оставшихся 9 VHL -мутантных опухолей (дополнительный рисунок S7B), с VHL — опухоль дикого типа, демонстрирующая низкие сигналы h4K27ac на суперэнхансере ZNF395 , сравнимые с нормой, соответствующей пациенту (дополнительная рис. S7C). В совокупности анализ путей энхансеров, истощенных в ≥2 клеточных линиях, выявил прямые эффекторы p53, передачу сигналов интегрин-связанной киназы и сети факторов транскрипции HIF1α в качестве пяти основных путей (дополнительная таблица S14), охватывающих такие гены, как EGFR (рис.4D), CCND1 (рис. 4E), ITGB3 (рис. 4F), VEGFA (рис. 4G), SLC2A1 (дополнительный рис. S7D) и HK2 (дополнительный рис. S7E) . Эти результаты подтверждают важную роль потери VHL в нарушении функции энхансера ccRCC, даже в присутствии других мутаций драйвера.
Интересно, что неконтролируемая кластеризация с использованием 1564 VHL -чувствительных полученных энхансеров отделила одну VHL -опухоль дикого типа (ID 75416923) от оставшихся 9 VHL -мутантных опухолей (дополнительный рисунок S7B), с VHL — опухоль дикого типа, демонстрирующая низкие сигналы h4K27ac на суперэнхансере ZNF395 , сравнимые с нормой, соответствующей пациенту (дополнительная рис. S7C). В совокупности анализ путей энхансеров, истощенных в ≥2 клеточных линиях, выявил прямые эффекторы p53, передачу сигналов интегрин-связанной киназы и сети факторов транскрипции HIF1α в качестве пяти основных путей (дополнительная таблица S14), охватывающих такие гены, как EGFR (рис.4D), CCND1 (рис. 4E), ITGB3 (рис. 4F), VEGFA (рис. 4G), SLC2A1 (дополнительный рис. S7D) и HK2 (дополнительный рис. S7E) . Эти результаты подтверждают важную роль потери VHL в нарушении функции энхансера ccRCC, даже в присутствии других мутаций драйвера.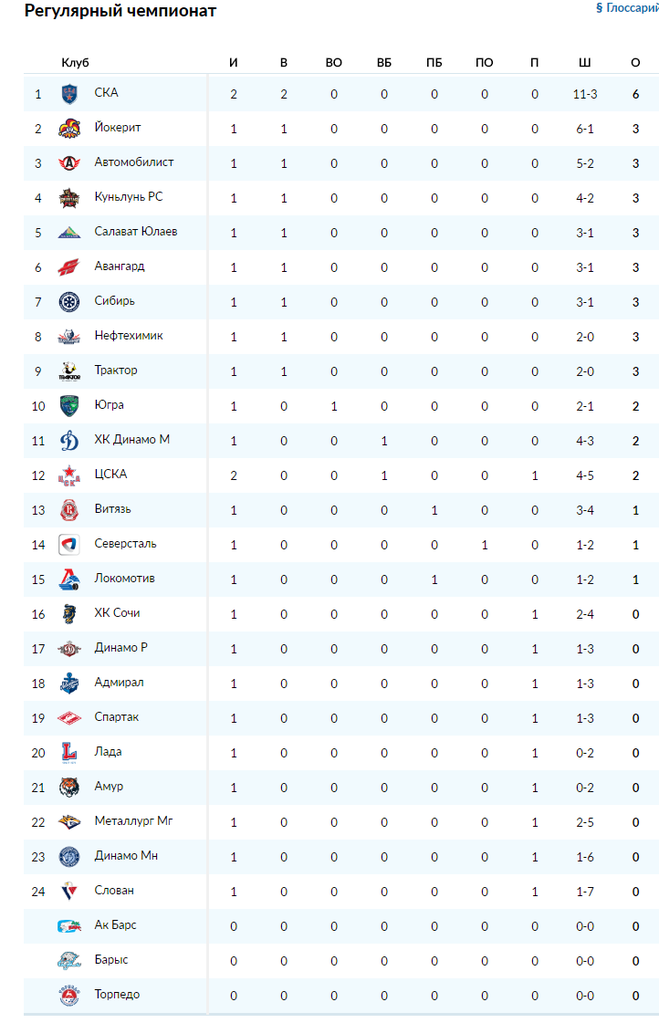
Мы также исследовали, были ли другие гистоновые метки изменены одновременно с метками h4K27ac. Мы обнаружили удивительно высокую степень корреляции между h4K27ac и h4K4me1 в ответ на восстановление VHL в обеих клетках 786-O ( r = 0.77, корреляция Пирсона, дополнительная рис. S7F) и 12364284 ячейки ( r = 0,61, корреляция Пирсона, дополнительная рис. S7G). Во всем мире энхансеры, демонстрирующие истощение h4K27ac, также испытывают сопутствующее истощение h4K4me1 (Fig. 4H). Затем мы исследовали, привело ли восстановление VHL к приобретению репрессивной метки h4K27me3. Несмотря на умеренную антикорреляцию h4K27ac и h4K27me3 (клетки 786-O: r = -0,28, корреляция Пирсона, дополнительный рисунок S7H; 12364284 клетки: r = -0.22, корреляция Пирсона, дополнительная рис. S7I), уровни h4K27me3 оставались низкими при полученных энхансерах даже после восстановления VHL (рис. 4H). Эти находки предполагают, что восстановление VHL может привести к потере идентичности энхансера за счет кодеплеции h4K27ac и h4K4me1, но не к формальному переходу в уравновешенное состояние энхансера, которое бы сохранило h4K4me1, но приобрело h4K27me3.
Гетеродимеры HIF2α-HIF1β обогащены VHL-чувствительными энхансерами
Мы стремились исследовать, какие факторы транскрипции могут опосредовать VHL-зависимое ремоделирование хроматина в полученных энхансерах.Начиная с первичного набора данных ccRCC, мы искали обогащение транс--регуляторов в приобретенных энхансерах по сравнению с потерянными энхансерами. Используя HOMER (65), мы обнаружили, что наиболее обогащенными мотивами были семейство AP1, семейство ETS и мотивы NF-κB–p65–Rel и HIF1α/2α (рис. 5A, полный список мотивов в дополнительной таблице S15). Для последующей проверки in vitro мы выбрали c-Jun в качестве репрезентативного члена семейства AP1 из-за его активации в ccRCC (66) и ETS1 в качестве представителя семейства ETS из-за его известного взаимодействия с HIF2α (67), но признаем, что другие члены семьи AP1 и семьи ETS могут играть роль в ccRCC.Иммуноблоттинг c-Jun, ETS1 и NF-κB-p65 показал переменную экспрессию белка как в нормальных, так и в опухолевых клеточных линиях, но экспрессия HIF1α и HIF2α ограничивалась только опухолевыми клетками (рис.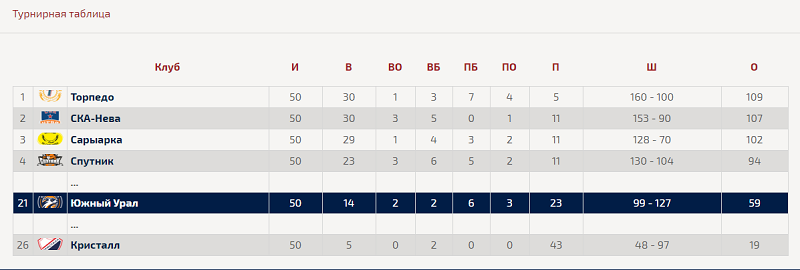 5B). HIF2α экспрессировался в более высокой доле клеточных линий ccRCC, чем HIF1α (фиг. 5B). Далее мы исследовали экспрессию генов этих транскрипционных факторов в когорте TCGA и обнаружили, что ETS1, RELA (субъединица NF-κB–p65) и HIF2α были значительно сверхэкспрессированы в опухолях по сравнению с нормальными тканями, с рядом опухолевых Паттерны экспрессии ассоциации, подобные вариациям линий ccRCC (дополнительная фиг.С8А).
5B). HIF2α экспрессировался в более высокой доле клеточных линий ccRCC, чем HIF1α (фиг. 5B). Далее мы исследовали экспрессию генов этих транскрипционных факторов в когорте TCGA и обнаружили, что ETS1, RELA (субъединица NF-κB–p65) и HIF2α были значительно сверхэкспрессированы в опухолях по сравнению с нормальными тканями, с рядом опухолевых Паттерны экспрессии ассоциации, подобные вариациям линий ccRCC (дополнительная фиг.С8А).
HIF2α обогащен энхансерами VHL -чувствительных опухолей. A, Анализ мотивов полученных энхансеров с использованием HOMER выявил значительное обогащение семейства AP1, семейства ETS, NF-κB и HIF1α/2α (гипергеометрический тест). Потерянные энхансеры использовали в качестве фона при поиске мотивов для идентификации опухолеспецифических факторов транскрипции. B, Экспрессия белков предполагаемых факторов транскрипции, обогащенная полученными энхансерами, в 9 линиях опухолевых клеток (T; 4 коммерческих клеточных линии и 5 клеточных линиях, полученных от пациентов) и 2 нормальных клеточных линиях (N). 2. Для дальнейшего изучения занятости хроматином этих факторов мы создали профили связывания ChIP-seq клеток c-Jun, ETS1 и NF-κB и повторно исследовали профили связывания HIF2α, HIF1α и HIF1β из предыдущей литературы (21, 30), все выполнено в ячейках 786-O.Следует отметить, что клетки 786-O утратили эндогенную экспрессию HIF1α из-за геномной делеции, HIF1α ChIP-seq была выполнена на клетках 786-O, подвергшихся генетическим манипуляциям для повторной экспрессии белка HIF1α (30). Чтобы определить, какие из этих транскрипционных факторов могут напрямую зависеть от VHL , мы затем сравнили экспрессию их белков в VHL -мутантных изогенных клеточных линиях с восстановлением дикого типа — VHL и без него.Как показано на рис. 5D, восстановление VHL последовательно подавляло экспрессию HIF2α как в 786-O, так и в 12364284 клеточных линиях, но уровни белка других факторов демонстрировали противоположные тенденции между двумя клеточными линиями, подразумевая, что среди шести исследованных факторов белок HIF2α экспрессия была наиболее зависимой от VHL. Действительно, подтверждая важную роль HIF2α в VHL-зависимом ремоделировании энхансера, только HIF2α и HIF1β были значительно обогащены энхансерами, демонстрирующими VHL-зависимое истощение h4K27ac (фиг. Напротив, HIF1α не был обогащен энхансерами, демонстрирующими истощение h4K27ac (фиг. 5E). Несмотря на общие с HIF2α многие сайты связывания, HIF1α преимущественно локализуется в проксимальных областях промотора, тогда как HIF2α часто занимает интроны и межгенные области в клетках 786-O (дополнительная рис.S8B), что согласуется с промоторной оккупацией HIF1α и энхансер-центрической оккупацией HIF2α (рис. 5F). Полученные энхансеры демонстрировали занятость HIF2α в два раза больше, чем опухолеспецифические промоторы ( P < 1 × 10 -16 , тест пропорций) в клетках 786-O, что позволяет предположить, что HIF2α может играть большую роль в регуляции энхансеров, чем промоторы. Чтобы распространить эти данные о характере занятости HIF1α и HIF2α на систему, которая экспрессирует эндогенные уровни обоих факторов, мы затем выполнили HIF1α и HIF2α ChIP-seq в 40 2 клетках ccRCC, которые обильно коэкспрессируют обе субъединицы HIFα (рис. 2 HIF1α демонстрировал преимущественную занятость в проксимальных областях промотора, тогда как большая часть HIF2α была обнаружена в дистальных областях (интроны и дистальные межгенные области; дополнительная рис. S8C). Более высокая доля сайтов связывания HIF1α, перекрывающихся с полученными промоторами, чем HIF2α (68% HIF1α против 41% HIF2α, P = 0,002, тест пропорций; рис. 5G). И наоборот, более высокая доля сайтов связывания HIF2α, перекрывающихся с полученными энхансерами, чем у HIF1α (29% HIF1α против51% HIF2α, P < 2,2 × 10 91 243 -16 91 244 , тест пропорций). Преимущественная занятость HIF2α в энхансерах была дополнительно подтверждена его более высоким обогащением в энхансерах, демонстрирующих истощение h4K27ac после восстановления VHL , чем HIF1α (фиг. 5H). Конкретные примеры чувствительных к VHL энхансеров, связанных исключительно с HIF2α, но не с HIF1α, включают энхансер вблизи UBR4 (фиг. ACHN представляет собой папиллярную клеточную линию ПКР. C, ChIP-seq подтвердили обогащение факторов транскрипции (TF) в приобретенных энхансерах по сравнению с утраченными энхансерами. D, Экспрессия белков факторов транскрипции показана в клетках 786-O и 12364284 с VHL дикого типа и без него. E, Связывание фактора транскрипции в VHL -чувствительных полученных энхансерах показывает обогащение HIF2α и HIF1β в энхансерах с истощением h4K27ac (красный) по сравнению с областями с обогащением h4K27ac (черный) после восстановления VHL .Данные F, ChIP-seq показывают распределение экзогенного связывания HIF1α и эндогенного HIF2α с измененными промоторами и энхансерами в клетках 786-O, которые были генетически сконструированы для сверхэкспрессии HIF1α. G, ChIP-seq показывает распределение эндогенного связывания HIF1α и HIF2α с измененными промоторами и энхансерами в клетках 40
ACHN представляет собой папиллярную клеточную линию ПКР. C, ChIP-seq подтвердили обогащение факторов транскрипции (TF) в приобретенных энхансерах по сравнению с утраченными энхансерами. D, Экспрессия белков факторов транскрипции показана в клетках 786-O и 12364284 с VHL дикого типа и без него. E, Связывание фактора транскрипции в VHL -чувствительных полученных энхансерах показывает обогащение HIF2α и HIF1β в энхансерах с истощением h4K27ac (красный) по сравнению с областями с обогащением h4K27ac (черный) после восстановления VHL .Данные F, ChIP-seq показывают распределение экзогенного связывания HIF1α и эндогенного HIF2α с измененными промоторами и энхансерами в клетках 786-O, которые были генетически сконструированы для сверхэкспрессии HIF1α. G, ChIP-seq показывает распределение эндогенного связывания HIF1α и HIF2α с измененными промоторами и энхансерами в клетках 40 H, Связывание фактора транскрипции в VHL-чувствительных энхансерах показывает более высокое обогащение HIF2α, чем HIF1α в энхансерах с истощением h4K27ac после восстановления VHL (красный) по сравнению с областями с обогащением h4K27ac после восстановления VHL (черный). I, Пример чувствительного к VHL энхансера вблизи UBR4 , связывающего только HIF2α, но не связывающего HIF1α. J, Пример VHL-чувствительного суперэнхансера, близкого к CMIP , связывающего только HIF2α, но не связывающего HIF1α.
H, Связывание фактора транскрипции в VHL-чувствительных энхансерах показывает более высокое обогащение HIF2α, чем HIF1α в энхансерах с истощением h4K27ac после восстановления VHL (красный) по сравнению с областями с обогащением h4K27ac после восстановления VHL (черный). I, Пример чувствительного к VHL энхансера вблизи UBR4 , связывающего только HIF2α, но не связывающего HIF1α. J, Пример VHL-чувствительного суперэнхансера, близкого к CMIP , связывающего только HIF2α, но не связывающего HIF1α. Результаты ChIP-seq показали, что все шесть транскрипционных факторов проявляли повышенную занятость в приобретенных энхансерах по сравнению с потерянными энхансерами, подтверждая предсказания HOMER (Fig. 5C).
Результаты ChIP-seq показали, что все шесть транскрипционных факторов проявляли повышенную занятость в приобретенных энхансерах по сравнению с потерянными энхансерами, подтверждая предсказания HOMER (Fig. 5C). 5Е). Более того, среди всех известных мотивов в базе данных HOMER HIF2α был наиболее обогащенным мотивом в VHL-чувствительных энхансерах, демонстрирующих истощение h4K27ac ( P = 1 × 10 -11 ; дополнительная таблица S16).
5Е). Более того, среди всех известных мотивов в базе данных HOMER HIF2α был наиболее обогащенным мотивом в VHL-чувствительных энхансерах, демонстрирующих истощение h4K27ac ( P = 1 × 10 -11 ; дополнительная таблица S16). 5Б). Подобно 786-O, в клетках 40
5Б). Подобно 786-O, в клетках 40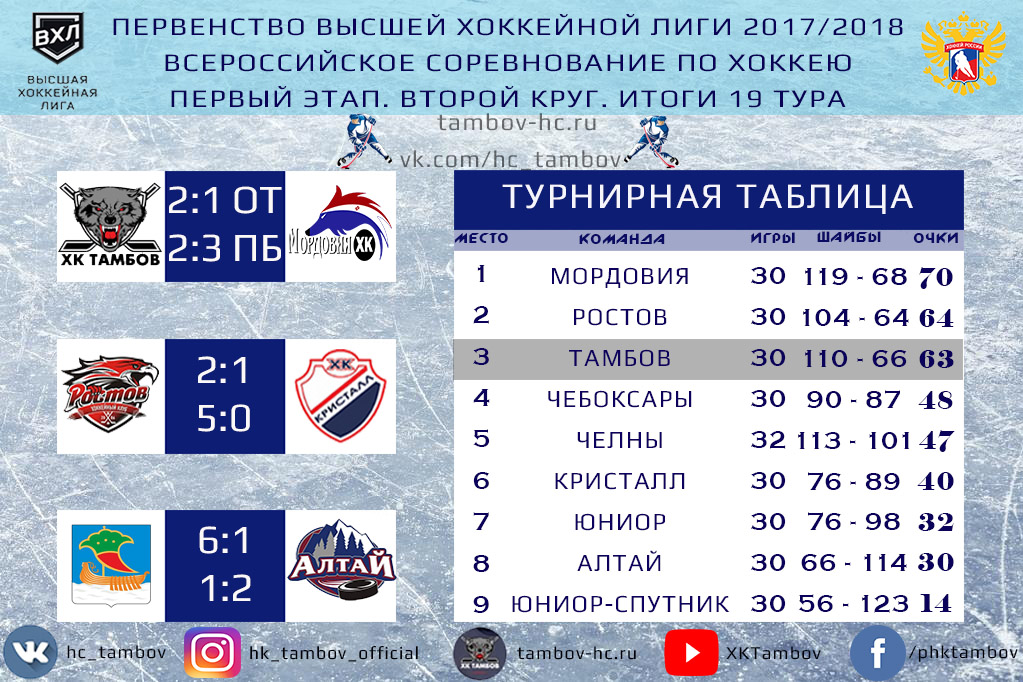 5I) и суперэнхансер вблизи CMIP (фиг. 5J). Таким образом, даже в клетках ccRCC, коэкспрессирующих HIF1α/HIF2α, эти результаты свидетельствуют о том, что HIF2α играет большую роль в VHL-опосредованном ремоделировании энхансера, чем HIF1α.
5I) и суперэнхансер вблизи CMIP (фиг. 5J). Таким образом, даже в клетках ccRCC, коэкспрессирующих HIF1α/HIF2α, эти результаты свидетельствуют о том, что HIF2α играет большую роль в VHL-опосредованном ремоделировании энхансера, чем HIF1α. Энхансеры, связанные с HIF2α–HIF1β, модулируют экспрессию генов О-клетки с нокдауном, опосредованным
HIF2α siRNA, и проанализированы корреляции между нокдауном HIF2α siRNA и восстановлением VHL . При оценке по всем генам корреляция была низкой ( r = 0.1, P = 5,2 × 10 −31 ) между нокдауном HIF2α и восстановлением VHL . Важно, однако, что эта корреляция увеличилась до 0,23 ( P = 5,8 × 10 -14 ) для генов вблизи сайтов связывания HIF2α (рис. 6А). Сходные результаты были получены на эпигеномном уровне, где для полученных энхансеров корреляция была низкой и составляла 0,06 для всех полученных энхансеров ( P = 1,9 × 10 -5 ), но значительно возросла до 0,37 ( P = 9.5 × 10 -8 ) у HIF2α-связанных энхансеров (рис. 6B) и у суперэнхансеров увеличилось с 0,089 ( P = 0,0025) до 0,25 ( P = 0,00054) у HIF2α-связанных суперэнхансеров (рис. 6C) . В качестве визуального примера, сигналы h4K27ac в суперэнхансере ZNF395 были уменьшены после восстановления VHL или нокдауна HIF2α , что сопровождалось снижением экспрессии гена ZNF395 (рис. 6D). Проверка с помощью RT-qPCR показала, что нокдаун HIF2α siRNA снижает экспрессию VEGFA, SLC2A1 и ZNF395 в степени, сравнимой с восстановлением VHL (фиг.6Е). Снижение люциферазной репортерной активности энхансерных элементов также соответствовало нокдауну HIF2α siRNA и восстановлению VHL (фиг. 6F).
Связанные энхансеры HIF2α–HIF1β модулируют экспрессию генов. A, Корреляция Пирсона изменения экспрессии генов либо после восстановления VHL , либо после нокдауна HIF2α siRNA во всех генах или генах, смежных с сайтами связывания HIF2α. EV, пустой вектор. B, Корреляция Пирсона изменений h4K27ac после восстановления VHL и нокдауна HIF2α siRNA либо во всех полученных энхансерах, либо в энхансерах, связанных с HIF2α, смежных с сайтами связывания. C, Корреляция Пирсона изменений h4K27ac после восстановления VHL и нокдауна HIF2α siRNA либо во всех полученных суперэнхансерах, либо в HIF2α-связанных суперэнхансерах, расположенных рядом с сайтами связывания. D, Изменения сигналов RNA-seq и h4K27ac ChIP-seq после восстановления VHL или нокдауна HIF2α siRNA на суперэнхансере ZNF395 (SE), вместе с профилями связывания факторов транскрипции, обогащенных энхансерами. E, Как восстановление VHL , так и нокдаун HIF2α siRNA снижают экспрессию генов с энхансерами, связанными с HIF2α, в клетках 786-O.*, P < 0,05, двусторонний тест t . F, Как восстановление VHL , так и нокдаун миРНК HIF2α снижают энхансерную активность, измеренную с помощью репортерного анализа люциферазы в клетках 786-O. *, P < 0,05, двусторонний тест t . G, RT-qPCR измерение экспрессии ZNF395 в четырех клонах дикого типа (черный) и четырех клонах с энхансером ZNF395 , истощенным CRISPR (красный). Истощенная область имеет самое высокое связывание HIF2α с суперэнхансером ZNF395 в клетках 786-O (удаленная область указана в D ).*, P < 0,05, двусторонний тест t .
Мы стремились установить причинно-следственную связь между энхансерами, связанными с HIF2α, и контролем экспрессии генов. Мы выполнили CRISPR-опосредованное геномное истощение области энхансера ZNF395 с самым высоким пиком HIF2α (рис. 6G). Все четыре клона с гомозиготным делетированным энхансером ZNF395 последовательно подавляли экспрессию ZNF395 по сравнению с клонами с интактным энхансером ( P < 0.05), предоставляя доказательства того, что экспрессия ZNF395 эпигенетически контролируется этим связанным с HIF2α-HIF1β энхансером (Fig. 6G). Взятые вместе, эти результаты показывают, что этот HIF2α, вероятно, является важным медиатором VHL-управляемого ремоделирования энхансера.
Восстановление VHL снижает рекрутинг p300, но сохраняет взаимодействие промотор-энхансерНаконец, мы попытались выяснить, почему восстановление VHL вызвало снижение уровней h4K27ac. Предыдущие анализы pulldown показали, что как HIF2α, так и HIF1β могут взаимодействовать с ацетилтрансферазой гистонов p300 (68–71).Действительно, p300 часто маркирует энхансеры (43) и, как полагают, рекрутируется тканеспецифичными факторами транскрипции (72). Однако профили хроматина р300 ранее не были установлены в клеточных линиях рака почки, поэтому вклад р300 в формирование энхансеров при скПКР остается неясным. Поэтому мы выполнили p300 ChIP-seq в клетках 786-O и подтвердили его обогащение полученными энхансерами по сравнению с потерянными энхансерами (рис. 7А). Сравнение p300 ChIP-seq с HIF2α ChIP-seq показало удивительно высокую степень перекрытия между HIF2α и p300 (96%), даже больше, чем у HIF2α и HIF1β (89%; рис.7В и С). Напротив, другие факторы транскрипции, такие как c-Jun, ETS1 и NF-κB, не проявляли такой высокой степени перекрытия (≤60%; рис. 7B).
Рисунок 7.Восстановление VHL снижает рекрутирование p300, но сохраняет взаимодействия промотор-энхансер. A, . Обогащение связывания p300 в приобретенных и потерянных энхансерах на основе ChIP-seq. B, Процент перекрытия между HIF2α и другими факторами транскрипции. C, ChIP-seq профили связывания HIF2α и p300. D, . Экспрессия белка р300 с VHL и без него в клетках 786-0 и 12364284, измеренная иммуноблоттингом. EV, пустой вектор. E, ChIP-qPCR связывания p300 на энхансерах с восстановлением VHL и без него в клетках 786-O. NC, участки отрицательного контроля. F, ChIP-qPCR связывания p300 на энхансерах с нокдауном HIF2α siRNA и без него в клетках 786-O. NC, участки отрицательного контроля. G, Корреляция взаимодействий энхансеров, измеренная с помощью Capture-C (об/мин, число прочтений на миллион) между клетками 786-O с восстановлением и без восстановления VHL как на VHL -чувствительных, так и на не- VHL -чувствительных энхансерах. H, Capture-C показывает, что взаимодействия энхансер-промотор VEGFA сохраняются даже после восстановления VHL . Е, энхансер; П, промоутер. I, Схематическая аберрация энхансера, вызванная VHL, при скПКР.
Мы сравнили связывание p300 с опухолевыми энхансерами с VHL и без него. Несмотря на повышенные уровни белка р300 в клетках 786-О после восстановления VHL (рис. 7D), связывание р300 снижалось для всех четырех исследованных энхансеров (рис. 7Е). Истощение HIF2α путем нокдауна siRNA также снижало рекрутирование p300 (фиг.7F), предполагая, что потеря HIF2α может мешать рекрутированию p300.
Мы исследовали, нарушают ли восстановление VHL и последующая потеря связывания p300 взаимодействия промотора и энхансера. Мы выполнили Capture-C областей энхансера в парных клеточных линиях 786-O с восстановлением VHL и без него. Неожиданно, взаимодействия Capture-C показали относительно высокую корреляцию между VHL -дефицитными и VHL -восстановленными клетками 786-O в VHL-чувствительных областях ( r = 0.74, корреляция Пирсона), даже выше, чем корреляции, наблюдаемые в областях, не реагирующих на VHL ( r = 0,57, корреляция Пирсона; рис. 7G). В качестве визуального примера, взаимодействия между промотором VEGFA и энхансером оставались интактными даже после восстановления VHL (рис. 7H), что указывает на то, что потеря активности энхансера, вероятно, недостаточна для диссоциации взаимодействий промотор-энхансер. Более того, многие из этих промоторов-энхансеров были клоноспецифичными; например, взаимодействие между энхансером SLC2A1 и его промотором не было обнаружено в KATOIII, клеточной линии рака желудка (дополнительная рис.С9). Следовательно, взаимодействия промотора и энхансера часто предсуществуют в клетках почек, часто тканеспецифическим образом.
Обсуждение
Понимание эпигеномных изменений и их генетического происхождения может выявить новые механизмы заболевания (34), уязвимости (73, 74) и терапевтические стратегии (75–77). Благодаря всестороннему профилированию модификаций гистонов в первичных парах норма-опухоль и клеточных линиях мы создали сборник промоторов и энхансеров, связанных с ccRCC. Наше исследование демонстрирует, что наиболее частое мутационное событие зПКР — инактивация VHL — приводит к полногеномному ремоделированию энхансеров и суперэнхансеров, что непосредственно придает отличительные признаки зПКР, включая ангиогенез и метаболическое перепрограммирование. ZNF395 , эпигенетически контролируемый VHL-чувствительным суперэнхансером, стал важным регулятором онкогенеза ccRCC.
В нашей работе есть три основных достижения. Во-первых, насколько нам известно, это наиболее полный атлас профилей гистонов при скПКР, который, вероятно, предоставит бесценный ресурс в области скПКР. Используя мультиплексированные интерактомные данные с высоким разрешением (Capture-C, ссылка 56) и корреляцию экспрессии h4K27ac, мы свели к минимуму неопределенность в назначении энхансеров и дополнительно подтвердили зависимость энхансеров от статуса VHL/HIF с помощью репортерных анализов.Во-вторых, используя изогенные клеточные линии, мы показываем, что потеря VHL вносит значительный вклад в ремоделирование энхансера. Несмотря на то, что другая мутация в зПКР, SETD2 , может опосредовать широко распространенное увеличение доступности хроматина (46) и гипометилирование ДНК (78), ее относительно низкая частота мутаций около 10% во всех опухолях зПКР (78) не может объяснить эпигенетические аномалии в подавляющее большинство SETD2 – опухоли дикого типа. Наконец, исследование соматически измененных суперэнхансеров позволило нам идентифицировать основной регулятор, имеющий решающее значение для патогенеза ccRCC, ZNF395 .Несмотря на то, что ранее сообщалось о гиперэкспрессии ZNF395 при ccRCC (79–81) и его близость к суперэнхансеру была отмечена независимо (42), наше исследование является первым, в котором точно определен специфический VHL-зависимый энхансер, необходимый для экспрессии ZNF395 и чтобы показать незаменимую функциональную роль ZNF395 в онкогенезе ccRCC in vitro и in vivo .
Наши данные свидетельствуют о том, что механически потеря VHL стабилизирует оккупацию HIF2α в опухолеспецифических энхансерах, которые, в свою очередь, привлекают гистон-ацетилтрансферазу p300 (28, 82) для поддержания ацетилирования h4K27, повышая экспрессию генов, специфичных для ccRCC, таких как ZNF395 (Инжир.7И). Восстановление VHL дикого типа приводило к кодеплеции меток h4K27ac и h4K4me1 и, таким образом, к отмене идентичности активного энхансера в ассоциированных с опухолью энхансерах. По сравнению с HIF1α, ориентированным на промотор, HIF2α преимущественно обнаруживается в энхансерах, что указывает на ключевое различие между HIF1α и HIF2α. Мы также обнаружили, что нокдаун HIF2α siRNA специфически ослабляет активность HIF2α-связанных энхансеров/суперэнхансеров. Интересно, что большинство взаимодействий промотор-энхансер оставались в значительной степени неизменными в результате статуса VHL , предполагая, что эти связи промотор-энхансер в значительной степени стабильны и сформированы заранее.Это согласуется с недавним сообщением, демонстрирующим, что взаимодействия промотора и энхансера остаются в значительной степени неизменными между нормоксией и гипоксией (29). Наше исследование, демонстрирующее влияние VHL на ремоделирование хроматина, также предполагает, что другие раковые гены с высокой мутационной пенетрантностью в зависимости от типа опухоли, такие как BRAF при меланоме (83) и APC при раке толстой кишки (84), также могут модифицировать клеточные эпигеномы, чтобы вызывать обширные, но специфичные для заболевания клеточные изменения, несмотря на то, что эти гены не являются классическими модификаторами хроматина.
Помимо VHL , мутации в других генах, таких как PBRM1, SETD2, ARID1A, SMARCA4, JARID1C и KDM6A/UTX , были зарегистрированы при ccRCC (47, 85), и они, вероятно, усиливают VHL 1. Основные транскрипционные эффекты (86), способствующие гетерогенности фенотипов заболевания (87) и моделей прогрессирования (88). На примере клеток 786-О по крайней мере две другие мутации в этих клетках могут оказывать прямое влияние на хроматин — MLL3 (стр.A3902G) и мутации TP53 с приобретением функции (p.R248W). MLL3 , гистон 3 лизин 4 метилтрансфераза, непосредственно отвечает за формирование энхансерной метки h4K4me1 (89, 90) и играет критическую роль в регуляции энхансера (91). Мутанты TP53 с приобретением функции также аберрантно связываются с хроматином, особенно рядом с метилтрансферазами MLL1 и MLL2 , потенциально способствуя росту опухоли посредством дерегуляции хроматина (92). Помимо мутаций, также известно, что структурные варианты изменяют энхансеры посредством захвата энхансера (93) или увеличения числа копий (94) при других видах рака.Учитывая множество драйверных и свидетельских мутаций при ccRCC, маловероятно, что только VHL может объяснить все эпигеномные изменения, наблюдаемые в этом типе опухоли. Тем не менее, путем интеграции данных по нескольким клеточным линиям скПКР наши данные позволяют предположить, что инактивация VHL , вероятно, составляет почти треть (32%) всех полученных энхансерных областей, подтверждая его роль в качестве доминирующего фактора эпигенетических аномалий при скПКР, несмотря на наличие других генетических изменений.
Наши эпигенетические карты содержат источник как хорошо проверенных, так и неохарактеризованных мишеней, которые могут способствовать онкогенезу скПКР. Мы обнаружили значительное увеличение энхансеров вокруг хорошо охарактеризованных мишеней, связанных с гипоксией (ссылка 95; VEGFA, CXCR4 , ссылка 96; HK2 ), мембранных переносчиков, опосредованных SLC ( SLC2A1, SLC2A2, SLC38A1 , ссылка 97). ), семейство SLC16A (98) и адипогенез ( PLIN2 , ссылки 51, 99). Новые мишени, обнаруженные в этом исследовании, включают SMPDL3A , который может быть еще одним важным онкогеном, специфичным для ccRCC, учитывая его роль в метаболизме липидов и холестерина (61, 100).Гены, связанные с потерянными суперэнхансерами, которые можно было идентифицировать только с парами нормальная-опухоль, связаны с потенциальными опухолевыми супрессорами ( EHF, MAL, GCOM1 и HOXB9 ), которые требуют дальнейшего изучения.
Одним из важных результатов этого эпигеномного исследования является онкогенная потребность в ZNF395 при скПКР. ZNF395 также известен как HDBP2 (101) или фактор связывания папилломавируса (PBF; ссылка 102). ZNF395 необходим для дифференцировки мезенхимальных стволовых клеток в адипоциты путем взаимодействия с PPARγ2 для стимуляции адипогенеза (103).Было показано, что ZNF395 связывается с промоторами гена Хантингтона (101) и генов, индуцированных интерфероном, и вызывает активацию генов, связанных с раком ( MACC1, PEG10, CALCOCO1 и MEF2C ; ссылка 104). и проангиогенные хемокины, включая IL6 и IL8 в условиях гипоксии (105). В будущих исследованиях еще предстоит выяснить точный механизм, способствующий туморогенной роли ZNF395 .
Ландшафты энхансеров, описанные в этом исследовании, имеют последствия не только для ccRCC.Плохо перфузируемое ядро опухоли делает гипоксию характерной чертой практически всех солидных опухолей (106). Клетки MCF7 в условиях гипоксии (но не нормоксии) имеют сходные профили h4K27ac с 786-O (29). Хотя ZNF395 высоко экспрессируется при ccRCC, его низкая базальная экспрессия может усиливаться при гипоксии при других типах рака, включая глиобластому и рак кожи (104, 105). Нацеливание на ZNF395 или его нижележащие эффекторы в будущих исследованиях может иметь терапевтическое значение как для ccRCC, так и для других гипоксических солидных злокачественных новообразований.Прямое нацеливание на ZNF395 с использованием противораковой вакцины на основе пептидов проходит испытания фазы I у пациентов с саркомой (107–109), что открывает возможность использования иммунотерапии для нацеливания на внеклеточные фрагменты основных ядерных регуляторов. Наше исследование предполагает, что проведение аналогичного исследования при скПКР может иметь смысл. Более того, учитывая недавний прогресс в нацеливании на факторы транскрипции с использованием различных методов, включая малые молекулы и сшитые пептиды (110–112), ингибиторы ZNF395 могут стать важным терапевтическим шагом в лечении скПКР.
Методы
Информация для пациентов
Свежезамороженные нормальные опухолевые ткани были получены из случаев нефрэктомии с одобрения комитетов по этике проведения исследований в учреждениях и с согласия пациентов в соответствии с протоколом 2010/735/B институционального наблюдательного совета. Нормальные ткани брали из участков, удаленных от опухоли. Подробную информацию о пациенте см. в дополнительной таблице S1.
Клеточные линии
Коммерческие клеточные линии (786-O, A-498, HK2 и PCS-400) были приобретены в ATCC.Линии клеток поддерживали в RPMI (Invitrogen) с 10% FBS, за исключением первичных эпителиальных клеток проксимальных канальцев почек, PCS-400, которые содержали в базальной среде для эпителиальных клеток почек (ATCC). Аутентификация клеточной линии была выполнена с помощью анализа коротких тандемных повторов (STR) (Институт рака Сингапура) в 2015 году по общедоступным профилям STR. Анализ на микоплазму проводили с использованием набора для ПЦР MycoSensor (Stratagene).
Создание опухолевых клеточных линий из первичных опухолей
Опухолевые клетки отделяли от первичных опухолей с помощью коллагеназы, высевали и поддерживали в RPMI с 10% FBS.При слиянии от 80% до 90% клетки пассировали в соотношении 1:3. Культивируемые клетки считали успешно иммортализованными после 60 пассажей. Правильное спаривание опухолевых тканей и клеточных линий было достигнуто путем сравнения процентной идентичности однонуклеотидных полиморфизмов (SNP) на основе направленного секвенирования. Все пары опухолевых клеточных линий показали идентичность > 90%, тогда как перетасовка пар показала идентичность < 80%. Опухоли и клеточные линии из 12364284 и 40
2 показали одинаковые мутации VHL , но ткань 86049102 (названная 86049102T) является мутантной VHL , тогда как родственная клеточная линия 86049102 (названная 86049102L-type2HL-2HL-wild-9002L) представляет собой
Стабильный
VHL Восстановление в линиях ccRCCКлетки 786-O (WT2, VHL + ) и клетки 786-O (RC3, VHL − 91 были любезно предоставлены Michael) Университет Торонто). Стабильная трансдукция VHL выполнялась в клетках A-498, 12364284 и 40
2 следующим образом: HA- VHL плазмида wt-pBabe-puro (подарок доктора Уильяма Келина, Институт рака Даны Фарбер; плазмида Addgene # 19234 ) трансфицировали в клетки PlatA (RV-102, Cell Biolabs) в количестве 2 мкг ДНК/лунку 6-луночного планшета с использованием липофектамина 3000 (LifeTechnologies).Смену среды проводили через 10-16 часов после трансфекции. Супернатант от клеток PlatA, содержащих ретровирусы, собирали через 48 часов и добавляли к клеткам ccRCC, которые затем отбирали пуромицином в течение 3 дней после трансдукции.
Histone Nano-ChIP-seq
Nano-ChIP-seq выполняли, как описано ранее (113) с небольшими изменениями. Свежезамороженные раковые и нормальные ткани рассекали с помощью лезвия бритвы, чтобы получить примерно 5 мг ткани. Ткани фиксировали в 1% формальдегиде в течение 10 минут при комнатной температуре.Фиксацию останавливали добавлением глицина до конечной концентрации 125 нмоль/л. Кусочки ткани промывали 3 раза буфером TBSE. Измельченные ткани лизировали в 100 мкл лизирующего буфера и обрабатывали ультразвуком в течение 16 циклов (30 с, 30 с без) с использованием Bioruptor (Diagenode). Использовали следующие антитела: h4K27ac (ab4729, Abcam), h4K4me3 (07-473, Millipore), h4K4me1 (ab8895, Abcam) и h4K27me3 (07-449, Millipore). Общий объем иммунопреципитации составил 1 мл, количество использованного антитела — 2 мкг.Введенную ДНК предварительно очищали с помощью Dynabeads с белком G (Life Technologies) в течение 1 часа при 4°C, а затем инкубировали с гранулами, конъюгированными с антителами, с белком G в течение ночи при 4°C. Гранулы промывали 3 раза холодным промывочным буфером. После выделения ChIP и введенной ДНК проводили полногеномную амплификацию с использованием набора WGA4 (Sigma-Aldrich) и праймеров BpmI-WGA. Амплифицированную ДНК расщепляли с помощью BpmI [New England Biolabs (NEB)]. После этого 30 нг амплифицированной ДНК использовали с набором реагентов для подготовки библиотеки NEBNext ChIP-seq (NEB).ChIP-seq в клеточных линиях выполняли с использованием того же протокола Nano-ChIP-seq, описанного выше, но с 1 × 10 6 клеток. Каждая библиотека была секвенирована со средней глубиной от 20 до 30 миллионов необработанных чтений на HiSeq2500 с использованием 101-битных считываний с одного конца.
Анализ последовательностей гистоновых чипов
Теги секвенирования были сопоставлены с эталонным геномом человека (hg19) с использованием выравнивателя Burrows-Wheeler (BWA-mem; ссылка 114; версия 0.7.10). Прочтения были обрезаны на 10 п.н. спереди и сзади, чтобы получить 81 п.н.Для последующего анализа использовались только чтения с mapQ > 10 и с удалением дубликатов с помощью rmdup. Значимые пики были названы с использованием CCAT ( P <0,05; ссылка 115). Силу и качество иммунопреципитации оценивали с помощью CHANCE (116).
Фактор транскрипции ChIP-seq
Для каждого фактора транскрипции 3 × 10 7 клеток сшивали 1% формальдегидом в течение 10 минут при комнатной температуре и останавливали добавлением глицина до конечной концентрации 125 нмоль/л.Хроматин экстрагировали и обрабатывали ультразвуком до ~500 п.н. (ячейка Vibra, SONICS). Для иммунопреципитации хроматина использовали следующие антитела: c-Jun (sc-1694, Santa Cruz Biotechnology), NF-κB p65 (sc-372, Santa Cruz Biotechnology), ETS1 (sc-350, Santa Cruz Biotechnology), HIF1α (610959 , BD Biosciences), HIF2α (NB100-122, Novus Bio) и p300 (sc-585, Santa Cruz Biotechnology). Общий объем иммунопреципитации составил 1,5 мл, а количество использованного антитела — 15 мкг. Вводимую ДНК предварительно очищали с помощью белков G Dynabeads (LifeTechnologies) в течение 2 часов при 4°C, а затем инкубировали с конъюгированными антителами белками G в течение ночи при 4°C.Гранулы промывали 6 раз промывочным буфером при комнатной температуре. Не менее 10 нг ДНК использовали с набором реагентов для подготовки библиотеки NEBNext ChIP-seq (NEB). Каждая библиотека была секвенирована со средней глубиной от 30 до 50 миллионов прочтений на HiSeq2500 с использованием односторонних прочтений 101 п.н.
Capture-C
Capture-C выполняли, как описано ранее (56). Вкратце, 1 × 10 7 клеток сшивали 2% формальдегидом с последующим лизисом, гомогенизацией, расщеплением DpnII, лигированием и удалением сшивания.ДНК обрабатывали ультразвуком с использованием Covaris до размера от 150 до 200 п.н. для получения ДНК, подходящей для захвата олигонуклеотидов. Всего для подготовки библиотеки секвенирования (NEB) использовали 3 мкг расщепленной ДНК. Энхансерные последовательности были дважды захвачены путем гибридизации с биотинилированными олигонуклеотидами (IDT) и обогащены Dynabeads (Life Technologies). Захваченная ДНК была секвенирована со средней глубиной 2 миллиона прочтений на зонд на платформе HiSeq Illumina с использованием прочтений парных концов длиной 150 п.н.
Анализ Capture-C и назначение гена
Предварительная обработка необработанных считываний была выполнена для удаления последовательностей адаптера (trim_galore, http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), а перекрывающиеся чтения были объединены с помощью FLASH (117). Чтобы добиться сопоставления короткого чтения с эталонным геномом hg19, полученные предварительно обработанные чтения были затем in silico расщеплены с помощью DpnII и выровнены с использованием Bowtie (с использованием параметров p1, m2, best и strata). Выровненные чтения обрабатывали с использованием анализатора Capture-C (118) для (i) удаления дубликатов ПЦР; (ii) классифицировать подфрагменты как «захват», если они содержались внутри фрагмента захвата, «исключение близости», если они находились в пределах 1 КБ по обе стороны от фрагмента захвата, или «репортер», если они находились за пределами «захвата» и регионы «исключения близости»; и (iii) нормализовать количество прочтений на 100 000 взаимодействий в формате bigwig.Мы дополнительно использовали пакет r3Cseq (119) на фрагментах захвата и репортера, чтобы определить значительные взаимодействия точки зрения на масштабированном фоне ( q -значение <0,05). Назначение генов определяется перекрытием значительных пиков Capture-C с генами с началом и концом, определенными GENCODE v19. Взаимодействия были построены с использованием Epigenome Gateway v40.0.
Идентификация областей с дифференциальным обогащением
Значимые пики h4K27ac, названные CCAT, были объединены во всех образцах нормальной опухоли.То же самое было выполнено с данными ChIP-seq h4K4me1 и h4K4me3. TSS были основаны на GENCODE v19. Промоторы определяли как области перекрытия между h4K27ac и h4K4me3, а также перекрывающиеся на ±2,0 т.п.о. вокруг TSS. Энхансеры определяли как области перекрытия между h4K27ac и h4K4me1, но не перекрывающиеся с промоторами. Чтобы свести к минимуму загрязнение стромы, мы провели дальнейшую фильтрацию с использованием данных клеточных линий, при этом энхансеры и промоторы, не перекрывающиеся с пиками h4K27ac ни в одной из клеточных линий, были отброшены.Файлы Wiggle с размером окна 50 п.н. были сгенерированы с использованием MEDIP (120) из файлов bam. Входной вычитаемый сигнал для каждой области промотора или энхансера был рассчитан с использованием bigWigAverageOverBed, чтобы получить количество прочтений на тысячу оснований на миллион (RPKM). RPKM h4K27ac, h4K4me1 и h4K4me3 ChIP-seq из промоторов и энхансеров корректировали с учетом эффектов партии с помощью Combat. Опухолеспецифические области определяли как области, которые имеют кратную разницу ≥2 и разницу 0,5 RPKM от нормальной ткани, соответствующей пациенту.Нормальные области были определены как области, которые имеют кратную разницу ≤0,5 и разницу -0,5 RPKM от соответствующих областей в опухолях, соответствующих пациенту. Рекуррентно увеличенные области определялись как увеличение у ≥5/10 пациентов и отсутствие потери ни у одного пациента. Периодически потерянные области определялись как потеря у ≥5/10 пациентов и отсутствие увеличения ни у одного пациента. Статистическое тестирование для каждой цис -регуляторной области проводили с использованием парных тестов t с поправкой Бенджамини-Хохберга.Дифференциальные области визуализировали с помощью NGSplot (121).
Идентификация областей суперэнхансеров
Области суперэнхансеров идентифицировали с использованием ROSE (ссылка 35; без промотора), используя области пиков h4K27ac, объединенные у всех пациентов (как нормальной, так и опухолевой ткани). Файлы Wiggle с размером окна 50 п.н. были сгенерированы с использованием MEDIP (120) из файлов bam. Входной вычитаемый сигнал для каждого суперэнхансера был рассчитан с использованием bigWigAverageOverBed (сумма считываний по покрытым базам).Области суперэнхансеров ранжировали по средней разнице сигналов h4K27ac ChIP-seq в норме и опухоли. Полученные суперэнхансеры определяли как области, которые имеют средние дифференциальные сигналы ChIP-seq h4K27ac >0. Потерянные суперэнхансеры были определены как области, которые имеют средние дифференциальные сигналы ChIP-seq h4K27ac <0.
Дополнительные методы можно найти в дополнительных методах.
Вклад авторов
Концепция и дизайн: X. Yao, J. Tan, B.C. Тан, Б.T. Teh, P. Tan
Разработка методологии: X. Yao, J. Tan, K.J. Лим, З. Ли, М. Син, Г. Виджая, Дж.О.Дж. Дэвис, Г. Лох, Б.К. Тан, С.Г. Розен, С. Ли, Д.Л. Сильвер, Дж. Р. Хьюз, Б. Т. Teh, P. Tan
Сбор данных (предоставление животных, приобретение и лечение пациентов, предоставление помещений и т. д.): X. Yao, J. Tan, Z. Li, D. Huang, M. Xing, Y.S. Чан, Дж.З. Цюй, С.Т. Тай, Ю.Н. Лам, Дж.Х. Хонг, А.П. Ли-Лим, К.З. He, C. Xu, S.S. Myint, B.C. Тан, С.Г. Розен, К.В.С. Ченг, К.Т.Е. Чанг, П.Х. Тан, А. Лежава, Б.Т. Teh
Анализ и интерпретация данных (например, статистический анализ, биостатистика, вычислительный анализ): X. Yao, J. Tan, K.J. Лим, В.Ф. Оои, П. Гуан, К.З. Он, Т. Нанди, А. Камра, С.Г. Розен, Б.Т. Teh, P. Tan
Написание, рецензирование и/или пересмотр рукописи: X. Yao, J. Tan, K.J. Лим, Т. Нанди, Б.Т. Teh, P. Tan
Административная, техническая или материальная поддержка (например, отчетность или организация данных, создание баз данных): X.Яо, Дж. Тан, К.Дж. Лим, Дж. Кох, З. Ли, Ю.С. Чан, С.Т. Тай, М.С.В. Нг, Дж.С. Лин, Т. Нанди, Г. Лох, Б.К. Тан, С.Г. Розен, К.Ю, И.Б.Х. Тан, А. Лежава, Г. Стегер, Б.Т. Teh, P. Tan
Руководство исследованием: X. Yao, S.G. Rozen, B.T. Teh, P. Tan
Другое (предоставил важные технические консультации и реагенты): J.Y. Goh
Различия в регуляции плотных контактов и морфологии клеток между мутациями VHL и подтипами заболевания | BMC Рак
Махер Э.Р., Изелиус Л., Йейтс Дж.Р., Литтлер М., Бенджамин С., Харрис Р., Сэмпсон Дж., Уильямс А., Фергюсон СМА, Мортон Н.: Болезнь фон Хиппеля-Линдау: генетическое исследование. J Med Genet. 1991, 28 (7): 443-447. 10.1136/JMG.28.7.443.
КАС Статья пабмед ПабМед Центральный Google Scholar
Kim WY, Kaelin WG: Роль мутации гена VHL в развитии рака человека. Дж. Клин Онкол. 2004, 22 (24): 4991-5004. 10.1200/JCO.2004.05.061.
КАС Статья пабмед Google Scholar
Латиф Ф., Тори К., Гнарра Дж., Яо М., Дах Ф.М., Оркатт М.Л., Стакхаус Т., Кузьмин И., Моди В., Гейл Л. и др.: Идентификация гена-супрессора опухоли при болезни фон Хиппеля-Линдау. Наука. 1993, 260 (5112): 1317-1320. 10.1126/научн.84.
КАС Статья пабмед Google Scholar
Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, Latif F, Liu S, Chen F, Duh FM и др.: Мутации гена-супрессора опухоли VHL при почечной карциноме.Нат Жене. 1994, 7 (1): 85-90. 10.1038/нг0594-85.
КАС Статья пабмед Google Scholar
Whaley JM, Naglich J, Gelbert L, Hsia YE, Lamiell JM, Green JS, Collins D, Neumann HP, Laidlaw J, Li FP и др.: Мутации зародышевой линии в опухоли фон Хиппеля-Линдау гена-супрессора сходны с соматическими аберрациями фон Хиппеля-Линдау при спорадическом почечно-клеточном раке. Am J Hum Genet. 1994, 55 (6): 1092-1102.
КАС пабмед ПабМед Центральный Google Scholar
Шуин Т., Кондо К., Торигоэ С., Кишида Т., Кубота Й., Хосака М., Нагасима Й., Китамура Х., Латиф Ф., Збар Б. и др.: Частые соматические мутации и потеря гетерозиготности гена-супрессора опухоли фон Хиппеля-Линдау при первичном почечно-клеточном раке человека. Рак рез. 1994, 54: 2852-2855.
КАС пабмед Google Scholar
Foster K, Prowse A, Berg van den A, Fleming S, Hulsbeek MM, Crossey PA, Richards FM, Cairns P, Affara NA, Ferguson-Smith MA, et al: Соматические мутации фон Хиппеля Ген-супрессор опухоли при болезни Линдау при несемейной светлоклеточной карциноме почки.Хум Мол Жене. 1994, 3: 2169-2173. 10.1093/hmg/3.12.2169.
КАС Статья пабмед Google Scholar
Lee JY, Dong SM, Park WS, Yoo NJ, Kim CS, Jang JJ, Chi JG, Zbar B, Lubensky IA, Linehan WM и др.: Потеря гетерозиготности и соматические мутации опухолевого супрессора VHL ген при спорадических гемангиобластомах мозжечка. Рак рез. 1998, 58 (3): 504-508.
КАС пабмед Google Scholar
Maher ER, Kaelin WG: болезнь Гиппеля-Линдау. Медицина (Балтимор). 1997, 76 (6): 381-391. 10.1097/00005792-199711000-00001.
КАС Статья Google Scholar
Kaelin WG: Молекулярная основа синдрома наследственного рака VHL. Нат Рев Рак. 2002, 2 (9): 673-682. 10.1038/nrc885.
КАС Статья пабмед Google Scholar
Schoenfeld A, Davidowitz EJ, Burk RD: второй основной нативный продукт гена фон Хиппеля-Линдау, инициированный из места начала внутренней трансляции, действует как супрессор опухоли.Proc Natl Acad Sci USA. 1998, 95 (15): 8817-8822. 10.1073/пнас.95.15.8817.
КАС Статья пабмед ПабМед Центральный Google Scholar
Iliopoulos O, Ohh M, Kaelin WG: pVHL19 представляет собой биологически активный продукт гена фон Хиппеля-Линдау, возникающий в результате инициации внутренней трансляции. Proc Natl Acad Sci USA. 1998, 95 (20): 11661-11666. 10.1073/пнас.95.20.11661.
КАС Статья пабмед ПабМед Центральный Google Scholar
Бланкеншип С., Наглич Дж.Г., Уэйли Дж.М., Сейзингер Б., Клей Н.: Альтернативный выбор инициирующего кодона дает биологически активный продукт гена фон Хиппеля Линдау с активностью супрессора опухоли. Онкоген. 1999, 18 (8): 1529-1535. 10.1038/sj.onc.1202473.
КАС Статья пабмед Google Scholar
Kibel A, Iliopoulos O, DeCaprio JA, Kaelin WG: Связывание белка-супрессора опухоли фон Хиппеля-Линдау с элонгином B и C.Наука. 1995, 269: 1444-1446. 10.1126/науч.7660130.
КАС Статья пабмед Google Scholar
Пауза А., Ли С., Уоррелл Р.А., Чен Д.Ю., Берджесс В.Х., Линехан В.М., Клауснер Р.Д.: Продукт гена опухолевого супрессора фон Хиппеля-Линдау образует стабильный комплекс с человеческим CUL-2, членом семейство белков Cdc53. Proc Natl Acad Sci USA. 1997, 94 (6): 2156-2161. 10.1073/пнас.94.6.2156.
КАС Статья пабмед ПабМед Центральный Google Scholar
Lonergan KM, Iliopoulos O, Ohh M, Kamura T, Conaway RC, Conaway JW, Kaelin WG: Регуляция индуцируемых гипоксией мРНК белком-супрессором опухоли фон Хиппеля-Линдау требует связывания с комплексами, содержащими элонгины B/C и Cul2. Мол Селл Биол. 1998, 18 (2): 732-741.
КАС Статья пабмед ПабМед Центральный Google Scholar
Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin WG, Elledge SJ, Conaway RC, et al: Rbx1, компонент опухолевого супрессорного комплекса VHL и Убиквитинлигаза SCF.Наука. 1999, 284 (5414): 657-661. 10.1126/наука.284.5414.657.
КАС Статья пабмед Google Scholar
Iwai K, Yamanaka K, Kamura T, Minato N, Conaway RC, Conaway JW, Klausner RD, Pause A: Идентификация белка-супрессора опухоли фон Хиппеля-Линдау как части активного комплекса убиквитинлигазы E3 . Proc Natl Acad Sci USA. 1999, 96 (22): 12436-12441. 10.1073/пнас.96.22.12436.
КАС Статья пабмед ПабМед Центральный Google Scholar
Листван Дж., Имберт Г., Вирбелауэр С., Гстайгер М., Крек В.: Белок-супрессор опухоли фон Хиппеля-Линдау является компонентом активности убиквитин-протеинлигазы Е3. Гены Дев. 1999, 13: 1822-1833. 10.1101/гад.14.13.1822.
КАС Статья пабмед ПабМед Центральный Google Scholar
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ: Белок-супрессор опухоли VHL нацелен на факторы, индуцируемые гипоксией, для кислородзависимых протеолиз.Природа. 1999, 399 (6733): 271-275. 10.1038/20459.
КАС Статья пабмед Google Scholar
Tanimoto K, Makino Y, Pereira T, Poellinger L: Механизм регуляции фактора-1 альфа, индуцируемого гипоксией, белком-супрессором опухоли фон Хиппеля-Линдау. Эмбо Дж. 2000, 19 (16): 4298-4309. 10.1093/emboj/19.16.4298.
КАС Статья пабмед ПабМед Центральный Google Scholar
Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH: Связывание и убиквитилирование фактора-альфа, индуцируемого гипоксией, белком-супрессором опухоли фон Хиппеля-Линдау. Дж. Биол. Хим. 2000, 275 (33): 25733-25741. 10.1074/jbc.M002740200.
КАС Статья пабмед Google Scholar
Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG: Убиквитинирование фактора, индуцируемого гипоксией, требует прямого связывания с бета-доменом фон Белок Хиппеля-Линдау.Nat Cell Biol. 2000, 2 (7): 423-427. 10.1038/35017054.
КАС Статья пабмед Google Scholar
Суфан Р.И., Джуэтт М.А., Ох М.: Роль белка-супрессора опухоли фон Хиппеля-Линдау и гипоксии при почечной светлоклеточной карциноме. Am J Physiol Renal Physiol. 2004, 287 (1): F1-6. 10.1152/айпренал.00424.2003.
КАС Статья пабмед Google Scholar
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG: HIF-альфа нацелен на опосредованное VHL разрушение путем гидроксилирования пролина: значение для определения O2. Наука. 2001, 292 (5516): 464-468. 10.1126/научн.1059817.
КАС Статья пабмед Google Scholar
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ и др.: Ориентация HIF-альфа на фон Хиппеля-Линдау комплекс убиквитилирования путем O2-регулируемого гидроксилирования пролила.Наука. 2001, 292 (5516): 468-472. 10.1126/научн.1059796.
КАС Статья пабмед Google Scholar
Yu F, White SB, Zhao Q, Lee FS: Связывание HIF-1альфа с VHL регулируется чувствительным к стимулу гидроксилированием пролина. Proc Natl Acad Sci USA. 2001, 98 (17): 9630-9635. 10.1073/пнас.181341498.
КАС Статья пабмед ПабМед Центральный Google Scholar
Збар Б., Кишида Т., Чен Ф., Шмидт Л., Махер Э.Р., Ричардс Ф.М., Кросси П.А., Вебстер А.Р., Аффара Н.А., Фергюсон-Смит М.А. и др.: Мутации зародышевой линии в гене болезни фон Хиппеля-Линдау (VHL) у семьи из Северной Америки, Европы и Японии. Хум Мутат. 1996, 8 (4): 348-357. 10.1002/(SICI) 1098-1004 (1996) 8:4<348::AID-HUMU8>3.0.CO;2-3.
КАС Статья пабмед Google Scholar
Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, Gnarra JR, Orcutt ML, Duh FM, Glenn G, et al: Мутации зародышевой линии в супрессоре опухоли при болезни фон Хиппеля-Линдау ген: корреляции с фенотипом.Хум Мутат. 1995, 5 (1): 66-75. 10.1002/гуму.1380050109.
КАС Статья пабмед Google Scholar
Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH, Ratcliffe PJ, Maher ER: Контрастные эффекты на регуляцию HIF-1alpha вызывающими заболевание мутациями pVHL коррелируют с моделями онкогенеза в von Болезнь Гиппеля-Линдау. Хум Мол Жене. 2001, 10 (10): 1029-1038. 10.1093/хмг/10.10.1029.
КАС Статья пабмед Google Scholar
Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M, Kaelin WG: белковые мутанты фон Хиппеля-Линдау, связанные с заболеванием VHL типа 2C, сохраняют способность подавлять HIF. Хум Мол Жене. 2001, 10 (10): 1019-1027. 10.1093/хмг/10.10.1019.
КАС Статья пабмед Google Scholar
Кнаут К., Бекс С., Джемт П., Бухбергер А. Риск почечно-клеточной карциномы при болезни Гиппеля-Линдау 2 типа коррелирует с дефектами стабильности pVHL и взаимодействий HIF-1альфа.Онкоген. 2006, 25 (3): 370-377.
КАС пабмед Google Scholar
Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, Farese RV, Freeman RS, Carter BD, Kaelin WG, et al: Апоптоз нейронов, связанный с генами пролилгидроксилазы EglN3 и семейной феохромоцитомы : отбраковка в связи с развитием и рак. Раковая клетка. 2005, 8 (2): 155-167. 10.1016/j.ccr.2005.06.015.
Артикул пабмед Google Scholar
Li L, Zhang L, Zhang X, Yan Q, Minamishima YA, Olumi AF, Mao M, Bartz S, Kaelin WG: Фактор, индуцируемый гипоксией, связан с дифференциальным риском рака почки, наблюдаемым с мутациями VHL типа 2A и типа 2B. Мол Селл Биол. 2007, 27 (15): 5381-5392. 10.1128/МКБ.00282-07.
КАС Статья пабмед ПабМед Центральный Google Scholar
Kondo K, Kim WY, Lechpammer M, Kaelin WG: Ингибирование HIF2-альфа достаточно для подавления роста опухоли с дефектом pVHL.PLoS биол. 2003, 1 (3): E83-10.1371/journal.pbio.0000083.
Артикул пабмед ПабМед Центральный Google Scholar
Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG: Ингибирование HIF необходимо для подавления опухоли белком фон Хиппеля-Линдау. Раковая клетка. 2002, 1 (3): 237-246. 10.1016/С1535-6108(02)00043-0.
КАС Статья пабмед Google Scholar
Zimmer M, Doucette D, Siddiqui N, Iliopoulos O: Ингибирование фактора, индуцируемого гипоксией, достаточно для подавления роста VHL-/-опухолей. Мол Рак Рез. 2004, 2 (2): 89-95.
КАС пабмед Google Scholar
Kim WY, Safran M, Buckley MR, Ebert BL, Glickman J, Bosenberg M, Regan M, Kaelin WG: Неспособность пролилгидроксилата индуцируемого гипоксией фактора альфа инактивация VHL in vivo. Эмбо Дж. 2006, 25 (19): 4650-4662.10.1038/sj.emboj.7601300.
КАС Статья пабмед ПабМед Центральный Google Scholar
Davidowitz EJ, Schoenfeld AR, Burk RD: VHL индуцирует дифференцировку почечных клеток и остановку роста за счет интеграции передачи сигналов между клетками и клетками и внеклеточным матриксом. Мол Селл Биол. 2001, 21 (3): 865-874. 10.1128/МКВ.21.3.865-874.2001.
КАС Статья пабмед ПабМед Центральный Google Scholar
Liebeau-Teillet B, Rak J, Jothy S, Iliopoulos O, Kaelin W, Kerbel RS: подавление роста, опосредованное генами фон Хиппеля-Линдау, и индукция дифференцировки клеток почечно-клеточной карциномы, выращенных в виде многоклеточных опухолевых сфероидов. Рак рез. 1998, 58 (21): 4957-4962.
КАС пабмед Google Scholar
Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, Gavin BJ, Kley N, Kaelin WG, Iliopoulos O: Белок-супрессор опухоли фон Хиппеля-Линдау необходим для правильного сборка внеклеточного матрикса фибронектина.Мол Ячейка. 1998, 1 (7): 959-968. 10.1016/С1097-2765(00)80096-9.
КАС Статья пабмед Google Scholar
Эстебан-Барраган М.А., Авила П., Альварес-Техадо М., Гутьеррес М.Д., Гарсия-Пардо А., Санчес-Мадрид Ф., Ландазури М.О.: Роль гена-супрессора опухоли фон Хиппеля-Линдау в формировании бета1 -интегрин фибриллярных спаек. Рак рез. 2002, 62 (10): 2929-2936.
КАС пабмед Google Scholar
Хергович А., Листван Дж., Барри Р., Балльшмитер П., Крек В.: Регуляция стабильности микротрубочек белком-супрессором опухоли фон Хиппеля-Линдау pVHL. Nat Cell Biol. 2003, 5 (1): 64-70. 10.1038/ncb899.
КАС Статья пабмед Google Scholar
Calzada MJ, Esteban MA, Feijoo-Cuaresma M, Castellanos MC, Naranjo-Suarez S, Temes E, Mendez F, Yanez-Mo M, Ohh M, Landazuri MO: белок-супрессор опухоли фон Хиппеля-Линдау регулирует сборка межклеточных соединений в клетках рака почки посредством индуцируемых гипоксией независимых от факторов механизмов.Рак рез. 2006, 66 (3): 1553-1560. 10.1158/0008-5472.CAN-05-3236.
КАС Статья пабмед Google Scholar
Lutz MS, Burk RD: Первичное образование ресничек требует функции гена von hippel-lindau в клетках почечного происхождения. Рак рез. 2006, 66 (14): 6903-6907. 10.1158/0008-5472.CAN-06-0501.
КАС Статья пабмед Google Scholar
Эстебан М.А., Хартен С.К., Тран М.Г., Максвелл П.Х.: Формирование первичных ресничек в почечном эпителии регулируется белком-супрессором опухоли фон Хиппеля-Линдау. J Am Soc Нефрол. 2006, 17 (7): 1801-1806. 10.1681/АСН.2006020181.
КАС Статья пабмед Google Scholar
Hughes MD, Kapllani E, Alexander AE, Burk RD, Schoenfeld AR: подавление HIF-2-альфа в отсутствие функциональной VHL недостаточно для дифференцировки почечных клеток.Раковая ячейка Интерн. 2007, 7: 13-10.1186/1475-2867-7-13.
Артикул пабмед ПабМед Центральный Google Scholar
Ji Q, Burk RD: Понижающая регуляция интегринов белком-супрессором опухоли фон Хиппеля-Линдау (VHL) не зависит от VHL-направленной деградации фактора альфа, индуцируемого гипоксией. Биохим Клеточная Биол. 2008, 86 (3): 227-234. 10.1139/О08-035.
КАС Статья пабмед ПабМед Центральный Google Scholar
Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ: Молекула, нацеленная на почечно-клеточную карциному с дефицитом VHL, которая вызывает аутофагию. Раковая клетка. 2008, 14 (1): 90-102. 10.1016/j.ccr.2008.06.004.
КАС Статья пабмед ПабМед Центральный Google Scholar
Эстебан М.А., Тран М.Г., Хартен С.К., Хилл П., Кастелланос М.С., Чандра А., Равал Р., О’Брайен Т.С., Максвелл П.Х.: Регуляция экспрессии Е-кадгерина с помощью VHL и фактора, индуцируемого гипоксией.Рак рез. 2006, 66 (7): 3567-3575. 10.1158/0008-5472.CAN-05-2670.
КАС Статья пабмед Google Scholar
Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, et al: Гипоксия способствует фиброгенезу in vivo посредством стимуляции HIF-1 эпителиально-мезенхимальный переход. Джей Клин Инвест. 2007, 117 (12): 3810-3820.
КАС пабмед ПабМед Центральный Google Scholar
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, eds: Current Protocols in Molecular Biology. 1994, Нью-Йорк: John Wiley and Sons
Schoenfeld AR, Davidowitz EJ, Burk RD: комплекс Elongin BC предотвращает деградацию продуктов генов-супрессоров опухолей фон Хиппеля-Линдау. Proc Natl Acad Sci USA. 2000, 97 (15): 8507-8512. 10.1073/пнас.97.15.8507.
КАС Статья пабмед ПабМед Центральный Google Scholar
Schoenfeld AR, Apgar S, Dolios G, Wang R, Aaronson SA: BRCA2 убиквитинируется in vivo и взаимодействует с USP11, деубиквитинирующим ферментом, который проявляет функцию выживания в клеточном ответе на повреждение ДНК. Мол Селл Биол. 2004, 24 (17): 7444-7455. 10.1128/МКБ.24.17.7444-7455.2004.
КАС Статья пабмед ПабМед Центральный Google Scholar
Brummelkamp TR, Bernards R, Agami R: Стабильное подавление канцерогенности с помощью вирусопосредованной РНК-интерференции.Раковая клетка. 2002, 2 (3): 243-247. 10.1016/С1535-6108(02)00122-8.
КАС Статья пабмед Google Scholar
Naviaux RK, Costanzi E, Haas M, Verma IM: Векторная система pCL: быстрое производство рекомбинантных ретровирусов с высоким титром без помощников. Дж Вирол. 1996, 70 (8): 5701-5705.
КАС пабмед ПабМед Центральный Google Scholar
Béroud C, Joly D, Gallou C, Staroz F, Orfanelli MT, Junien C: Программное обеспечение и база данных для анализа мутаций в гене VHL.Нуклеиновые Кислоты Res. 1998, 26 (1): 258-260.
Google Scholar
Кросси П.А., Ричардс Ф.М., Фостер К., Грин Дж.С., Проуз А., Латиф Ф., Лерман М.И., Збар Б., Аффара Н.А., Фергюсон-Смит М.А. и др.: Идентификация внутригенных мутаций в гене фон Хиппеля. Ген-супрессор опухоли при болезни Линдау и корреляция с фенотипом заболевания. Хум Мол Жене. 1994, 3: 1303-1308. 10.1093/hmg/3.8.1303.
КАС Статья пабмед Google Scholar
Stolle C, Glenn G, Zbar B, Humphrey JS, Choyke P, Walther M, Pack S, Hurley K, Andrey C, Klausner R и др.: Улучшенное обнаружение мутаций зародышевой линии в гене-супрессоре опухоли при болезни фон Хиппеля-Линдау. Хум Мутат. 1998, 12 (6): 417-423. 10.1002/(SICI) 1098-1004 (1998) 12:6<417::AID-HUMU8>3.0.CO;2-K.
КАС Статья пабмед Google Scholar
Браух Х., Кишида Т., Главац Д., Чен Ф., Пауш Ф., Хофлер Х., Латиф Ф., Лерман М.И., Збар Б., Нойманн Х.П.: Болезнь фон Хиппеля-Линдау (ВХЛ) с феохромоцитомой в Шварцвальде регион Германии: свидетельство эффекта основателя.Хам Жене. 1995, 95 (5): 551-556. 10.1007/BF00223868.
КАС Статья пабмед Google Scholar
Glavac D, Neumann HP, Wittke C, Jaenig H, Masek O, Streicher T, Pausch F, Engelhardt D, Plate KH, Hofler H, et al: Мутации в гене супрессора опухоли VHL и связанные с ними поражения в семей с болезнью фон Гиппеля-Линдау из Центральной Европы. Хам Жене. 1996, 98 (3): 271-280. 10.1007/s0043206.
КАС Статья пабмед Google Scholar
Chen F, Slife L, Kishida T, Mulvihill J, Tisherman SE, Zbar B: Корреляция генотип-фенотип при болезни фон Хиппеля-Линдау: идентификация мутации, связанной с VHL типа 2A. J Med Genet. 1996, 33 (8): 716-717. 10.1136/JMG.33.8.716.
КАС Статья пабмед ПабМед Центральный Google Scholar
Bradley JF, Collins DL, Schimke RN, Parrott HN, Rothberg PG: два разных фенотипа, вызванные двумя разными миссенс-мутациями в одном и том же кодоне гена VHL.Am J Med Genet. 1999, 87 (2): 163-167. 10.1002/(SICI)1096-8628(199
)87:2<163::AID-AJMG7>3.0.CO;2-A.
КАС Статья пабмед Google Scholar
Ольшванг С., Ричард С., Буассон С., Жиро С., Лоран-Пуиг П., Реше Ф., Томас Г.: Профиль мутаций зародышевой линии гена VHL при болезни фон Хиппеля-Линдау и спорадической гемангиобластоме. Хум Мутат. 1998, 12 (6): 424-430. 10.1002/(SICI)1098-1004(1998)12:6<424::AID-HUMU9>3.0,СО; 2-Н.
КАС Статья пабмед Google Scholar
Wu Y, Nishio H, Lee MJ, Ayaki H, Hayashi A, Ooba T, Ogawa T, Sumino K: Молекулярно-генетический анализ и скрининг мутаций гена VHL в японской семье с болезнью фон Гиппеля-Линдау . Коби J Med Sci. 2000, 46 (4): 147-153.
КАС пабмед Google Scholar
Кросси П.А., Фостер К., Ричардс Ф.М., Фиппс М.Е., Латиф Ф., Тори К., Джонс М.Х., Бентли Э., Кумар Р., Лерман М.И. и др.: Молекулярно-генетические исследования механизма онкогенеза у фон Хиппеля — Болезнь Линдау: анализ потери аллелей в опухолях VHL.Хам Жене. 1994, 93 (1): 53-58. 10.1007/BF00218913.
КАС Статья пабмед Google Scholar
Ritter MM, Frilling A, Crossey PA, Hoppner W, Maher ER, Mulligan L, Ponder BA, Engelhardt D: изолированная семейная феохромоцитома как вариант болезни фон Гиппеля-Линдау. J Clin Endocrinol Metab. 1996, 81 (3): 1035-1037. 10.1210/jc.81.3.1035.
КАС пабмед Google Scholar
Bindra RS, Vasselli JR, Stearman R, Linehan WM, Klausner RD: VHL-опосредованная регуляция гипоксии циклина D1 в клетках почечной карциномы. Рак рез. 2002, 62 (11): 3014-3019.
КАС пабмед Google Scholar
Zatyka M, da Silva NF, Clifford SC, Morris MR, Wiesener MS, Eckardt KU, Houlston RS, Richards FM, Latif F, Maher ER: Идентификация циклина D1 и других новых мишеней для фон Хиппель- Ген-супрессор опухоли Линдау с помощью анализа массива экспрессии и исследования генотипа циклина D1 как модификатора болезни фон Гиппеля-Линдау.Рак рез. 2002, 62 (13): 3803-3811.
КАС пабмед Google Scholar
Пауза А., Ли С., Лонерган К.М., Клауснер Р.Д.: Ген супрессора опухоли фон Хиппеля-Линдау необходим для выхода из клеточного цикла после удаления сыворотки. Proc Natl Acad Sci USA. 1998, 95 (3): 993-998. 10.1073/пнас.95.3.993.
КАС Статья пабмед ПабМед Центральный Google Scholar
Schoenfeld AR, Parris T, Eisenberger A, Davidowitz EJ, De Leon M, Talasazan F, Devarajan P, Burk RD: Ген-супрессор опухоли фон Хиппеля-Линдау защищает клетки от УФ-опосредованного апоптоза. Онкоген. 2000, 19 (51): 5851-5857. 10.1038/sj.onc.1203985.
КАС Статья пабмед Google Scholar
Moustakas A, Heldin CH: Сигнальные сети, направляющие эпителиально-мезенхимальные переходы во время эмбриогенеза и прогрессирования рака.Онкологические науки. 2007, 98 (10): 1512-1520. 10.1111/j.1349-7006.2007.00550.х.
КАС Статья пабмед Google Scholar
Evans AJ, Russell RC, Roche O, Burry TN, Fish JE, Chow VW, Kim WY, Saravanan A, Maynard MA, Gervais ML, et al: VHL способствует транскрипции Е2-бокс-зависимого E-кадгерина с помощью HIF-опосредованная регуляция SIP1 и snail. Мол Селл Биол. 2007, 27 (1): 157-169. 10.1128/МКБ.00892-06.
КАС Статья пабмед Google Scholar
Harten SK, Shukla D, Barod R, Hergovich A, Balda MS, Matter K, Esteban MA, Maxwell PH: Регуляция плотных контактов почечного эпителия с помощью гена-супрессора опухоли фон Хиппеля-Линдау включает окклюдин и клаудин 1 и не зависит от E -кадгерин. Мол Биол Селл. 2009, 20 (3): 1089-1101. 10.1091/mbc.E08-06-0566.
КАС Статья пабмед ПабМед Центральный Google Scholar
Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, Sellers WR: Регуляция прогрессирования G1 белком-супрессором опухоли PTEN связана с ингибированием пути фосфатидилинозитол-3-киназы/Akt .Proc Natl Acad Sci USA. 1999, 96 (5): 2110-2115. 10.1073/пнас.96.5.2110.
КАС Статья пабмед ПабМед Центральный Google Scholar
Bojarski C, Weiske J, Schoneberg T, Schroder W, Mankertz J, Schulzke JD, Florian P, Fromm M, Tauber R, Huber O: Специфические судьбы белков плотных контактов в апоптотических эпителиальных клетках. Дж. Клеточные науки. 2004, 117 (часть 10): 2097–2107. 10.1242/JCS.01071.
КАС Статья пабмед Google Scholar
Гороспе М., Иган Дж. М., Збар Б., Лерман М., Гейл Л., Кузьмин И., Холбрук Н. Дж.: Защитная функция белка фон Хиппель-Линдау против нарушения процессинга белка в клетках почечной карциномы. Мол Селл Биол. 1999, 19 (2): 1289-1300.
КАС Статья пабмед ПабМед Центральный Google Scholar
Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ: Сравнительные свойства фактора, индуцируемого гипоксией 1 (HIF-1) и HIF-2 при почечно-клеточной карциноме, ассоциированной с фон Хиппелем-Линдау.Мол Селл Биол. 2005, 25 (13): 5675-5686. 10.1128/МКБ.25.13.5675-5686.2005.
КАС Статья пабмед ПабМед Центральный Google Scholar
Rankin EB, Tomaszewski JE, Haase VH: Развитие почечной кисты у мышей с условной инактивацией опухолевого супрессора фон Хиппеля-Линдау. Рак рез. 2006, 66 (5): 2576-2583. 10.1158/0008-5472.CAN-05-3241.
КАС Статья пабмед ПабМед Центральный Google Scholar
Young AP, Schlisio S, Minamishima YA, Zhang Q, Li L, Grisanzio C, Signoretti S, Kaelin WG: потеря VHL активирует независимую от HIF программу старения, опосредованную Rb и p400. Nat Cell Biol. 2008, 10 (3): 361-369. 10.1038/ncb1699.
КАС Статья пабмед Google Scholar
Цихлиас Дж., Капуста Л., Слингерланд Дж.: Прогностическое значение измененных ингибиторов циклинзависимой киназы при раке человека. Анну Рев Мед.1999, 50: 401-423. 10.1146/аннурев.мед.50.1.401.
КАС Статья пабмед Google Scholar
Slingerland J, Pagano M: Регуляция ингибитора cdk p27 и его дерегуляция при раке. J Cell Physiol. 2000, 183 (1): 10-17. 10.1002/(SICI)1097-4652(200004)183:1<10::AID-JCP2>3.0.CO;2-I.
КАС Статья пабмед Google Scholar
Sicinski P, Zacharek S, Kim C: Двойственность функции p27Kip1 в онкогенезе. Гены Дев. 2007, 21 (14): 1703-1706. 10.1101/гад.1583207.
КАС Статья пабмед Google Scholar
Stebbins CE, Kaelin WG, Pavletich NP: Структура комплекса VHL-ElonginC-ElonginB: последствия для функции супрессора опухоли VHL. Наука. 1999, 284 (5413): 455-461. 10.1126/научн.284.5413.455.
КАС Статья пабмед Google Scholar
Фельдман Д.Е., Туласираман В., Феррейра Р.Г., Фридман Дж.: Формирование комплекса супрессора опухоли VHL-элонгин до н.э. опосредовано шаперонином TRiC. Мол Ячейка. 1999, 4 (6): 1051-1061. 10.1016/С1097-2765(00)80233-6.
КАС Статья пабмед Google Scholar
Lee CM, Hickey MM, Sanford CA, McGuire CG, Cowey CL, Simon MC, Rathmell WK: Мутация гена VHL Type 2B снижает дозировку HIF in vitro и in vivo. Онкоген.2009, 28 (14): 1694-1705. 10.1038/onc.2009.12.
КАС Статья пабмед ПабМед Центральный Google Scholar
Окуда Х., Хираи С., Такаки Ю., Камада М., Баба М., Сакаи Н., Кишида Т., Канеко С., Яо М., Оно С. и др.: Прямое взаимодействие бета-домена опухолевого супрессора VHL белок с регуляторным доменом атипичных изотипов ПКС. Biochem Biophys Res Commun. 1999, 263 (2): 491-497. 10.1006/bbrc.1999.1347.
КАС Статья пабмед Google Scholar
Suzuki A, Yamanaka T, Hirose T, Manabe N, Mizuno K, Shimizu M, Akimoto K, Izumi Y, Ohnishi T, Ohno S: Атипичная протеинкиназа C участвует в эволюционно консервативном комплексе белков par и играет критическую роль в установление специфических для эпителия соединительных структур. Джей Селл Биол. 2001, 152 (6): 1183-1196. 10.1083/jcb.152.6.1183.
КАС Статья пабмед ПабМед Центральный Google Scholar
Xu J, Clark RA: Трехмерная решетка коллагена индуцирует активность протеинкиназы C-zeta: роль в экспрессии мРНК альфа2-интегрина и коллагеназы.Джей Селл Биол. 1997, 136 (2): 473-483. 10.1083/jcb.136.2.473.
КАС Статья пабмед ПабМед Центральный Google Scholar
Laudanna C, Mochly-Rosen D, Liron T, Constantin G, Butcher EC: Доказательства участия дзета-протеинкиназы C в интегрин-зависимой адгезии и хемотаксисе полиморфноядерных нейтрофилов. Дж. Биол. Хим. 1998, 273 (46): 30306-30315. 10.1074/jbc.273.46.30306.
КАС Статья пабмед Google Scholar
Guo W, Giancotti FG: передача сигналов интегрина во время прогрессирования опухоли. Nat Rev Mol Cell Biol. 2004, 5 (10): 816-826. 10.1038/nrm1490.
КАС Статья пабмед Google Scholar
Paraf F, Chauveau D, Chretien Y, Richard S, Grunfeld JP, Droz D: Поражения почек при болезни фон Хиппеля-Линдау: иммуногистохимическая экспрессия молекул дифференцировки нефрона, молекул адгезии и белков апоптоза. Гистопатология.2000, 36 (5): 457-465. 10.1046/j.1365-2559.2000.00857.х.
КАС Статья пабмед Google Scholar
Bluyssen HA, Lolkema MP, van Beest M, Boone M, Snijckers CM, Los M, Gebbink MF, Braam B, Holstege FC, Giles RH и др.: Фибронектин является независимой от гипоксии мишенью опухоли. глушитель ВХЛ. ФЭБС лат. 2004, 556 (1–3): 137–142. 10.1016/S0014-5793(03)01392-9.
КАС Статья пабмед Google Scholar
Комбинированная делеция Vhl и Kif3a ускоряет образование почечной кисты
Abstract
Считается, что подмножество семейных и спорадических светлоклеточных почечно-клеточных карцином (ccRCC) развивается из кистозных предшественников.Потеря функции гена-супрессора опухоли фон Хиппеля-Линдау ( VHL ) предрасполагает клетки почечного эпителия к потере первичной реснички в ответ на специфические сигналы. Поскольку первичная ресничка подавляет образование почечной кисты, потеря реснички может быть инициирующим событием в формировании скПКР. Чтобы проверить эту гипотезу, мы проанализировали последствия индуцируемой специфичной для почечного эпителия делеции Vhl вместе с удалением первичной реснички посредством делеции члена семейства кинезинов 3A ( Kif3a ) гена.Мы разработали подход к визуализации на основе микрокомпьютерной томографии, позволяющий проводить количественный лонгитюдный мониторинг кистозной нагрузки, показывая, что комбинированная потеря Vhl и Kif3a сокращает латентный период образования кист, увеличивает количество кист на почку и увеличивает общее количество кистозных образований. груз. В отличие от результатов других кистозных моделей, в кистах Kif3a мутантных мышей не наблюдалось накопления индуцируемого гипоксией фактора 1- α (HIF1 α ), а делеции Hif1a и Kif3a не наблюдалось. повлиять на развитие или прогрессирование кисты.Двойная мутация Vhl/Kif3a также увеличивала частоту кист, которые демонстрировали многослойный эпителиальный рост, что коррелировало с повышенной частотой неправильно ориентированных делений кистозных эпителиальных клеток. Эти результаты опровергают участие HIF1 α в стимулировании роста почечных кист и предполагают, что образование простых и атипичных почечных кист, которые напоминают поражения, предшествующие ccRCC, значительно ускоряются при комбинированной потере Vhl и первичной реснички.
Болезнь Гиппеля-Линдау (ВГЛ) представляет собой аутосомно-доминантное генетическое заболевание, вызываемое наследованием мутантного аллеля гена ВГЛ . Пациенты с болезнью VHL имеют повышенный риск развития почечных кист и двустороннего, многоочагового солидного или кистозно-светлоклеточного почечно-клеточного рака (ccRCC), при которых аллель VHL дикого типа неизменно теряется из-за соматической мутации или молчания гена. 1 В почках пациентов с заболеванием VHL развивается спектр VHL -дефицитных поражений, которые, вероятно, представляют собой поражения-предшественники солидных и кистозных ccRCC, включая небольшие очаги микро-ccRCC с солидным внешним видом, простые кисты, выстланные одним слоем пролиферирующие эпителиальные клетки и атипичные кисты, содержащие участки многослойного эпителиального роста, напоминающие очаги скПКР. 2 Эти данные свидетельствуют о том, что зПКР образует посредством кист-зависимых и кист-независимых путей у пациентов с заболеванием VHL. 3 Ген VHL также биаллельно инактивирован в >90% спорадических случаев скПКР. 4 Хотя большинство зПКР имеют солидную морфологию, примерно 5% являются кистозными с небольшими узелковыми скоплениями светлоклеточных опухолевых клеток внутри и между стенками кист, 5,6 что позволяет предположить, что спорадический зПКР также возникает через кисты- зависимый и цистнезависимый пути.
pVHL опосредует многочисленные биологические активности, в том числе нацеливание на α -субъединицы факторов транскрипции, индуцируемых гипоксией (HIF1 α , HIF2 α и HIF3 α ) для протеолитической стабилизации 4 и протеолитической деградации 7 7 сеть. 8 Эта последняя функция pVHL важна для регуляции первичной реснички, структуры на основе микротрубочек, которая функционирует как сенсорная органелла для многочисленных химических и механических стимулов. 9 Генетические мутации, нарушающие сигнальные функции или структуру первичной реснички, вызывают поликистоз почек. 10 Опухоли ccRCC человека демонстрируют чрезвычайно низкую частоту реснитчатых клеток 11 , а потеря функции VHL в иммортализованных почечных эпителиальных клетках или клеточных линиях ccRC ухудшает образование первичных ресничек. 12–14 Однако делеция Vhl в почечных эпителиальных клетках почки мыши или в культивируемых первичных клетках не вызывает потери первичной реснички, но повышает чувствительность клеток к потере первичной реснички в ответ на стимуляцию фосфоинозитида 3 сигнальный путь -киназы (PI3K) и инактивация киназы гликогенсинтазы 3 β . 15,16 Мутантные кистозные эпителиальные клетки VHL в почках пациентов с заболеванием VHL часто лишены первичной реснички и демонстрируют гиперактивацию сигнального пути PI3K и повышенные уровни фосфорилированной, неактивной киназы гликогенсинтазы 3 β . 15,16 Считается, что потеря реснички является вторичным событием, которое происходит в некоторых мутантных клетках VHL и действует как триггер для инициации образования кист. Комбинированная делеция Vhl и Pten вместе в клетках почечного эпителия мышей, но не одного гена по отдельности, вызывает почечные эпителиальные кисты со сниженной частотой реснитчатых эпителиальных клеток, что подтверждает эту гипотезу. 15 Однако было невозможно сделать вывод, является ли образование кист результатом исключительно потери реснички, совместных эффектов потери реснички плюс потеря Vhl или совместных эффектов потери реснички плюс потеря Vhl плюс активация пути PI3K. Поскольку у мышей Vhl/Pten развились различные доброкачественные новообразования в половых путях, которые потребовали ранней эвтаназии, 17 также не было возможности определить возраст мышей, чтобы определить, способны ли мутантные кистозные поражения Vhl прогрессировать с образованием ccRCC.Здесь мы обращаемся к некоторым из этих ограничений, создавая мышиную модель, допускающую индуцибельную делецию Vhl вместе с генетической абляцией первичных ресничек посредством делеции Kif3a , которая кодирует белковый компонент kinesin-II моторного комплекса микротрубочек. 18
Потеря функции pVHL вызывает стабилизацию HIF1 α и HIF2 α в эпителиальных клетках кистозных поражений у пациентов с болезнью VHL. 2 Недавние исследования показывают, что HIF1 α может играть более общую роль в кистозных поражениях, возникающих при генетических состояниях, отличных от заболевания VHL.HIF1 α , но не HIF2 α , стабилизируется в клетках кистозного эпителия из-за микросредовой гипоксии в крысиной модели поликистозной болезни почек и в тканях человека с поликистозной болезнью почек. 19 На основании двух моделей образования кист in vitro было высказано предположение, что активация Hif1 α способствует образованию кист. 20 Однако вопрос о том, способствует ли HIF1 α образованию или прогрессированию кистозных поражений, не проверялся на физиологически значимой кистозной модели.Мы проверили эту идею, удаляя Hif1a в модели образования кисты почки с делецией Kif3a .
Результаты. -Cre
ERT2 Tg/+ ; Kif3a fl/fl ; Vhl fl/fl ) и Kif3a/Hif1a (Ksp1.3-Cre ERT2 Tg/+ ; Kif3a 9f/42l 9124; Hif1a фл/фл ).Нетрансгенные однопометники (Ksp1.3-Cre ERT2 +/+ ) служили контролем. Шестинедельных мышей кормили пищей, содержащей тамоксифен, в течение 2 недель, чтобы вызвать активацию рекомбиназы Cre. Обработанные животные далее именуемые как Kif3a Δ / Δ , Kif3a Δ / Δ ; VHL Δ / Δ , или KIF3A δ / δ ; Hif1a δ / δ и элементы управления как KIF3A FL , KIF3A FL / FL ; VHL фл/фл или Kif3a фл/фл ; Hif1a фл/фл соответственно.Рекомбинационно-специфическая ПЦР геномной ДНК, выделенной из почек, показала, что все гены были удалены, как и ожидалось (рис. 1А). Хотя этот метод активации Cre приводит к некоторым различиям между животными в степени делеции гена (дополнительная фигура 1K), локус Kif3a рекомбинировался с одинаковой частотой в среднем у Kif3a Δ / Δ , Kif3a Δ / Δ ; VHL Δ / Δ и Kif3a Δ / Δ ; Hif1a Δ / Δ почки (рис. 1В).Специфическая для почечного эпителия делеция Vhl вызывает накопление в ядре HIF1 α 15 и Делеция Kif3a вызывает потерю первичных ресничек по оценке окрашивания ацетилированным тубулином, 18 обеспечивает два считывания, которые позволяют идентифицировать ген клетки. Эпителиальные клетки, выстилающие кисты в Kif3a Δ / Δ , Kif3a Δ / Δ ; VHL Δ / Δ , Или KIF3A δ / / δ ; Hif1a ; Hif1a ; / δ мыши неизменно не хватало первичных ресничек (рисунок 1С).HIF1 α Атомное окрашивание наблюдалось во всех цистических эпителиальных клетках в KIF3A Δ δ ; VHL δ / δ , но не в KIF3A δ / / δ Мутантные мыши (рисунок 1с) Проверка удаления VHL и показывая, что кисты в KIF3A Δ / δ мышей не гипоксические, в отличие от результатов полученный на модели поликистоза почек у крыс. 19 канальцы в Kif3a δ Δ / δ ; VHL δ / δ Мутантные почки, в которых односмысленные или несколько эпителиальные клетки были положительными для HIF1 α окрашивание (дополнительная фигура 1B) наблюдались с частотой примерно в 10 раз выше, чем количество кист. Точно так же часто наблюдались канальцы, в которых некоторые клетки демонстрировали повышенное окрашивание на Glut-1, HIF1 α -индуцируемый белок и маркер делеции Vhl , тогда как другие клетки были отрицательными на Glut-1.В этих канальцах 78% Glut-1-негативных клеток, но только 4% Glut-1-позитивных клеток демонстрировали первичную ресничку, демонстрируя кодецию Vhl и Kif3a в этих клетках (дополнительный рисунок 1, H и J ). Эти канальцы, содержащие мутантные клетки Vhl / Kif3a , не обнаруживали явных морфологических аномалий, что означает, что кисты не образовывались в течение 9 месяцев после делеции гена. Мы пришли к выводу, что частота делеции гена не является ограничивающим фактором в определении степени образования кисты.Окрашивание маркерами различных сегментов нефрона показало, что кисты развиваются из проксимальных канальцев, толстых восходящих петель Генле, дистальных извитых канальцев, собирательных трубочек и мочевого полюса клубочка (дополнительный рисунок 2), что соответствует картине экспрессии Ksp1. 3-Cre ERT2 трансген. 21 Рис. 1. Поколение мышейс индуцируемой делецией Kif3a, Kif3a/Vhl или Kif3a/Hif1a . (А) ПЦР геномной ДНК от почек KIF3A Δ /
δ и KIF3A FL / FL (TOP), KIF3A δ / δ ; VHL Δ 4 / δ и Kif3a fl / fl ; VHL FL (средний), и KIF3A δ / δ ; Мыши Hif1a Δ / Δ и Kif3a fl/fl ; Hif1a fl/fl (внизу).Были проведены реакции ПЦР с использованием праймеров, специфичных для аллелей floxed (fl) и рекомбинации ( Δ ), и показаны положения ожидаемых продуктов. Промоторная область рибосомного белка S6 (S6) служит внутренним контролем. (B) Количественный анализ ПЦР в реальном времени соотношения Kif3a рекомбинированных и флоксированных аллелей в почках от указанных генотипов. Среднее ± стандартное отклонение ( n = 3–7). (C) Представитель H & E, Anti-HiF1 α , и антиацетилированный тубулиновый окрашивание гистологически нормальных и кистозных (CY) областей Kif3a Δ / Δ , Kif3a δ / Δ ; VHL δ / δ , и Kif3a δ / δ ; Hif1a Δ / Δ почки.H&E, гематоксилин и эозин. Bar, 20 µ M.Для количественного мониторинга образования и прогрессирования кист мы разработали методику визуализации на основе микрокомпьютерной томографии ( µ CT), включающую внутрисосудистое введение контрастного вещества, которое концентрируется через систему почечных канальцев. , что позволяет визуализировать структуру почки. 22 Кисты либо не получают контрастное вещество, либо не могут его концентрировать и поэтому выглядят как темные области на µ КТ.Мышей визуализировали через 8, 16, 20, 24, 28, 32 и 36 недель после введения тамоксифена в пищу (рис. 2). Почки выделяли и брали плазму крови после последней временной точки. µ КТ не выявила образования кисты у контрольных животных, но развитие кист у всех мутантных генотипов. Сравнение мкм КТ-изображений с гистологическими срезами тех же почек показало, что подход мкм КТ точно отражает кистозную нагрузку (рис. 2). Kif3a Δ / Δ и Kif3a Δ / Δ ; Hif1a Δ / Δ животных показали умеренное кистозный фенотип Характеризуется развитием от 2 до 20 мелких и средних кист на почку.Хотя некоторые KIF3A δ / δ
; VHL δ / δ мышки показали видимый умеренный цистический фенотип, многие разрабатывали тяжелый фенотип и проявляют много более больших кист . Общая кистозная нагрузка была количественно оценена с помощью трехмерной реконструкции объемов всех кистозных образований во всей почке (рис. 3А). По сравнению с Kif3a Δ / Δ и Kif3a Δ / Δ ; Hif1a Δ / Δ мышей, Kif3a δ 4 / / δ ; VHL δ / δ мышей разработаны кисты раньше и с более высокой проникшей во всех временных точках (рис. 3б) и показали резко повышенное общее кистоз нагрузка (рис. 3C).Гистологический анализ выявил, что KIF3A δ / δ ; VHL δ / δ почек показал 4-кратное увеличение количества кисты по сравнению с KIF3A Δ / Δ и Kif3a Δ / Δ; Hif1a Δ / Δ почки (рис 3D).Общий объем почек, определенный с использованием КТ мкм , отлично коррелировал с массой почек (дополнительная фигура 3, A – C), что еще раз подтвердило точность подхода КТ мкм . Creatininine (Дополнительный рисунок 3D) и мочевины (Дополнительный рисунок 3e) в плазме крови показали незначительные тенденции к увеличению уровней в KIF3A δ / δ ; VHL δ / δ мыши. Поскольку значения для всех контрольных животных были в пределах нормы, выполнение ежемесячных циклов КТ µ в течение жизни животного не ухудшает функцию почек.Кроме того, Kif3a Δ / Δ ; Hif1a Δ / Δ и Kif3a Δ / Δ ; VHL У мышей Δ / Δ , которых кормили тамоксифеном, но не подвергали ежемесячной визуализации, развивались кисты, аналогичные тем, которые подвергались повторной визуализации (дополнительная фигура 4), что показывает, что повторная инъекция контрастного вещества и µ КТ не влияет на кистозный фенотип. Рисунок 2.Продольная визуализация прогрессирования кисты. Представитель μ CT Изображения дикого типа (A), KIF3A Δ / Δ (b), Kif3a δ / δ ; VHL Δ / Δ (C) и Kif3a Δ / Δ 90; Мыши Hif1a Δ / Δ (D), взятые через 8, 16, 28 и 36 недель после лечения, и соответствующий гистологический срез одной почки (36 недель) от каждой из изображенных мышей.Мышей оценивали визуально как имеющих умеренный или тяжелый кистозный фенотип, и показано количество животных каждого генотипа с каждой оценкой. H&E, гематоксилин и эозин. Полоса 1 мм на изображениях, окрашенных гематоксилин-эозином.
Рисунок 3.Количественная оценка кистовой нагрузки раскрывает повышенную кистообразное образование в KIF3A δ
δ ; VHL δ / δ почек. (А) COROAL μ CT Изображение и трехмерная реконструкция представителя KIF3A δ / δ , KIF3A , KIF3A / δ ; VH L Δ 4 / δ , и KIF3A δ / δ ; Hif1a δ / δ Почки в 36 недель после лечения.Почки (желтые) и кисты (красные) показаны в верхней части рентгеновского изображения. (B) Процент животных с кистами почек на µ КТ-изображениях в разные моменты времени после делеции гена. Kif3a Δ / Δ ( п = 10), Kif3a Δ / Δ ; VHL Δ / Δ ( N = 16), и KIF3A Δ / / δ ; Hif1a ; Hif1a ; Hif1a / δ ( N = 9).(C) Количественная оценка общего кистозного объема на почку. Данные представлены как среднее значение ± SEM для KIF3A Δ / δ ( N = 14), KIF3A δ / δ ; VHL Δ / Δ ( п = 30), а Kif3a Δ / Δ ; Hif1a Δ / Δ ( n =16).(D) Количество кист на срез почки, определенное путем анализа гистологических срезов по продольной средней линии почек. Данные представлены как среднее значение ± SEM для KIF3A Δ / δ ( N = 24), KIF3A δ / δ ; VHL δ / Δ ( п = 27), а Kif3a Δ / Δ ; Hif1a Δ / Δ ( n =17).* Р <0,05; *** P <0,001 (непарный тест t ).Чтобы определить ли образование киста на киф3а δ
δ ; VHL Δ / / δ мышей результаты из увеличения сотовой пролиферации, мы использовали Бромодексиуридин (Brdu Маркировка пролиферирующих ячеек в Kif3a δ / δ ; VHL δ / / δ Почки 3 недели после инициирования Tamoxifen Cynding и анализируемых канальцев, отображающие удаление гена мозаики с использованием иммуноокрашивания Glut-1 в качестве маркера делеции Vhl (рис. 4, A и B).Glut-1-положительные и Glut-1-отрицательные клетки в этих канальцах демонстрировали очень низкую скорость пролиферации (<0,5%), а Glut-1-положительные клетки показали немного более низкую скорость пролиферации (рис. 4C), что указывает на то, что повышенная пролиферация не Не легендарное образование киста в KIF3A Δ / δ ; VHL δ / δ мышей. Чтобы сравнить пролиферативный статус эпителиальных клеток в кистозных поражениях из разных генотипов, мы выбрали группы мышей, которые демонстрировали сопоставимый умеренный кистозный фенотип.Окрашивание Ki67 показало, что некоторые кисты содержали пролиферирующие клетки, а другие — нет (рис. 4D). Количественными не выявлено различий между Kif3a Д / Δ , Kif3a Δ / Δ ; Hif1a Δ / Δ , и Kif3a δ / / δ ; VHL ; VHL δ / δ мыши в терминах частоты кисты, которые содержали пролиферирующие клетки (рис. 4е) или частота Ki67-положительные клетки в пролиферирующих кистах (рис. 4F).Увеличенный цистический номер и общая кистозная нагрузка в KIF3A δ / Δ ; VHL δ / Δ мышей, по-видимому, приведет к увеличению частоты инициирования кисты в более ранние моменты времени, а не повышенная скорость пролиферации кистозных клеток. Рисунок 4.Различий между генотипами в скорости пролиферации кистозных клеток нет. (A и B) Примеры канальцев из KIF3A Δ / δ ; VHL ; VHL δ / δ мышей, в которых ячейка дикого типа (B) и клетка с дефицитом Vhl , отмеченная положительным окрашиванием Glut-1 (зеленый) (A), окрашивается положительно на BrdU (красный).(C) Процент Glut-1-положительных ( n = 2629) и Glut-1-отрицательных ( n = 2798) клеток, которые окрашиваются положительно на BrdU. Данные получены из анализа нескольких разделов от трех KIF3A δ /
δ ; VHL δ / δ мышей. (D) Примеры кист каждого генотипа, которые либо не содержат клеток, окрашивающихся на Ki67 (Ki67 —), либо содержат клетки, меченные Ki67 (Ki67 + ).(E) Процент кист, которые отображают клетки, меченные Ki67. Данные представлены как среднее значение ± SEM для KIF3A δ / Δ ( N = 54), KIF3A δ / δ ; VHL Δ / Δ ( п = 56), а Kif3a Δ / Δ ; Hif1a Δ / Δ ( n = 62).(F) Процент Ki67-положительных клеток на Ki67-положительную кисту. Данные представлены как среднее значение ± SEM для KIF3A δ / δ ( N = 36), KIF3A δ / δ ; VHL δ / Δ ( п = 37), а Kif3a Δ / Δ ; Hif1a Δ / Δ ( n = 36).Бар, 10 мк м в А и В; 20 мкм м в D.Большинство кист всех генотипов представляли собой простые кисты, выстланные одним слоем эпителия (рис. 5, A–C). В Kif3a Δ / Δ и Kif3a Δ / Δ ; Hif1a Δ / Δ мутантных мышей, 1,9% ( n =204) и 2,1% ( n =282) кист соответственно имели небольшие очаги многослойного или дезорганизованного эпителиального разрастания, выступавшие в просвет кисты (рис. 5, А и В).Напротив, в KIF3A δ /
δ ; VHL ; VHL δ / δ мышей, частота этих атипичных кист было в 4 раза выше (8,0%, n =914) (рис. 5В). Атипичные кисты также наблюдались у мышей, которые не подвергались ежемесячной визуализации (дополнительная фигура 4D), что указывает на то, что этот фенотип не является вторичным эффектом экспериментального протокола. Опухоли у этих мышей не наблюдались. Рисунок 5.KIF3A δ /
δ ; VHL ; VHL ; / δ MICE отображает Увеличение частот пропущенного деления эпителиальной клеток и атипичных кист. Представитель H & E-E-образные разделы простых кисты (слева) и атипичных кисты (справа) от Kif3a Δ / δ (A), KIF3A δ / δ ;Vhl Δ / Δ (Б), и Kif3a Δ /4 4 4 Δ
4
4; Hif1a Δ / Δ (C) мыши через 36 недель после делеции гена индуцировали у взрослых мышей.(D) Окрашивание антифосфогистоном h4 (красный) и DAPI (синий) нормальных (слева) и неправильно ориентированных (справа) анафаз. Локальную плоскость кистозного эпителия определяли, проводя линию через два соседних ядра, а плоскость анафазы определяли, проводя линию, параллельную средней линии делящихся хромосом, для расчета угла анафазы α . (E) Количественная оценка угла α из анафаз в Kif3a δ /
δ ( N = 41) и KIF3A δ 4 δ ; Vhl Δ / Δ ( n =42) кистозный эпителий.* P <0,05, тест Манна-Уитни U . (F) Примеры расщепленной каспазы 3-позитивные кисты эпителиальные клетки в KIF3A δ / Δ и Kif3a δ / δ ; VHL δ / Δ почки. Стрелка указывает на апоптозную клетку, лежащую над эпителиальным слоем. (G) Частота кисты в KIF3A δ / / ( N = 518), KIF3A δ / δ ; VHL δ / Δ ( п = 582), а Kif3a Δ / Δ ; Hif1a Δ / Δ ( п =542) почки, в которых обнаружены одна или несколько расщепленных каспаз-3-положительных кистозных эпителиальных клеток.H&E, гематоксилин и эозин; DAPI, 4′,6-диамидино-2-фенилиндол. Бар, 20 мк м в А–С; 10 мк м в D; 20 µ m у F.Делеция генов, подавляющих кисты, на ранних стадиях постнатального развития вызывает более тяжелый и быстроразвивающийся кистозный фенотип, чем делеция генов в почках взрослых. 21,23 Мы вводили кормящим самкам дозу тамоксифена, которая индуцировала ограниченное количество кист у кормящихся детенышей, и следили за этим с помощью КТ µ в течение 9 месяцев (дополнительная фигура 5).Делеция Vhl у мышей P2 не вызывала развития кист (дополнительная фигура 5). Окрашивание на HIF1 α и ацетилированный тубулин подтвердило функциональные эффекты делеции Kif3a и Vhl в соответствующих генотипах (дополнительная фигура 6). Этот метод делеции давал значительные различия в степени образования кист внутри и между пометами, что не позволяло проводить значимые количественные сравнения кистозного бремени. Морфологический анализ почек через 9 месяцев после делеции гена выявил фенотипы, сходные с делецией гена у взрослых, в отношении образования простых и атипичных кист и отсутствия опухолей.В Kif3a Δ / Δ и Kif3a Δ / Δ ; Hif1a Δ / Δ мутантных мышей, 3,3% ( n = 333) и 3,5% ( n = 692) кист, соответственно, содержали области атипичного эпителиального роста (дополнительная фигура 7), тогда как 9,3% ( n = 647) кист в Kif3a Δ / Δ ;Vhl Δ / Δ мыши.Эти результаты в совокупности показывают, что комбинированная мутация Vhl и Kif3a увеличивает частоту перехода в атипичные кисты.
Возникновению кисты в нескольких животных моделях потери функции первичных ресничек предшествует дезориентация делений эпителиальных клеток канальцев, что приводит к делению клеток поперек плоскости канальца и расширению канальца. 24 Образование многослойного кистозного эпителия может также включать неплоское деление эпителиальных клеток.Анафаза клетка в кистозной эпителии Kif3a Δ / Δ и Kif3a Δ / Δ ; VHL Δ / Δ мышей идентифицировали путем окрашивания на фосфогистон h4, а плоскость клеточного деления определяли путем измерения угла между плоскостью кистозного эпителия и линией, параллельной средней линии разделяющихся митотических массивов хромосом (рис. 5D).Anaphases в KIF3A δ
δ ; VHL δ / δ Кисты отображаются значительно большее распространение углов, чем анафазы в KIF3A / Δ кисты (рис. 5Е). Примечательно, 33% KIF3A δ / δ ; VHL ; VHL δ / δ анафазы были ориентированы больше из плоскости эпителия, чем в плоскости эпителия (угол <45°), тогда как только 8% кистозных анафаз Kif3a Δ / Δ были смещены.Мы пришли к выводу, что неправильно ориентированное клеточное деление может привести к потере клеточного контакта с базальной мембраной, вызывая гибель клеток. Последовательный с этой идеей, KIF3A δ / δ ; VHL δ / δ Cysts Чаще отображаются один или несколько ячеек, которые положительно окрашиваются для расщепления Caspase 3, маркером апоптоза, чем кист в Kif3a Δ / Δ и Kif3a Δ / Δ ; Hif1a Δ / Δ почек (рис. 5, F и G).Во многих случаях эти расщепленные каспаза-3-положительные клетки наблюдались над слоем кистозного эпителия, что согласуется с нашей моделью.Обсуждение
Ранее мы предположили, что потеря функции pVHL, стабилизирующей микротрубочки, в сочетании с вторичными сигнальными изменениями в пути PI3K приводит к неспособности эпителиальных клеток поддерживать первичную ресничку, вызывая образование предопухолевых кист у пациентов с заболеванием VHL. 3,15,16 Это исследование подтверждает эту гипотезу, но также предлагает модификацию исходной модели формирования кисты.Комбинированная делеция Vhl и Kif3a сокращает латентный период образования кисты и увеличивает общее количество кист и общую кистозную нагрузку по сравнению с делецией только Kif3a , утверждая, что потеря первичной реснички не является единственным инициирующим событием в формирование кисты, но эта потеря первичной реснички действует совместно с потерей функции pVHL, вызывая образование кисты. Поскольку считается, что простые кисты прогрессируют в атипичные кисты при кистзависимом пути образования скПКР, повышенная частота атипичных кист у Kif3a Δ / Δ ;Vhl 30 4 Δ 4 Δ мышей указывает на то, что потеря Vhl плюс потеря ресничек способствует прогрессированию ccRCC.Однако, поскольку эти поражения сохраняются у мышей в течение по крайней мере 6-9 месяцев без прогрессирования в опухоли, вполне вероятно, что дополнительные мутации, часто встречающиеся при скПКР, такие как PBRM1 , BAP1 , SETD2 или TP53 или PI3K изменения пути 4,25 необходимы для образования опухоли из этих атипичных кист. В соответствии с идеей о том, что потеря ресничек является ранним событием в формировании ccRCC, спорадические опухоли человека ccRCC демонстрируют очень низкую частоту реснитчатых клеток. 11 Хотя геномный анализ скПКР не выявил мутации генов, кодирующих структурные компоненты первичных ресничек, 4,25 активация пути PI3K является частым событием 4,25–29 и, как известно, вызывает потерю ресничек в VHL нулевых клеток. 15,16 Возможно, что другие генетические изменения, которые еще предстоит идентифицировать, также могут взаимодействовать с потерей VHL , вызывая потерю реснички. Интересно, что гомозиготная делеция Vhl в сочетании с гетерозиготной делецией Bap1 во время развития почки мыши вызывает образование простых и атипичных кист, а также опухолей. 30 Наблюдение того, что эти повреждения демонстрируют высокие уровни активации пути PI3K, предполагает, что исследование состояния первичных ресничек у этих мышей является оправданным.
Поскольку многие нормальные канальцы, содержащие делецию Vhl и Kif3a , не образуют цист в течение 9 месяцев после делеции гена, очевидно, что потеря Vhl вместе с потерей первичных ресничек не приводит автоматически к образованию кист. Клеточная пролиферация после повреждения нефронов вызывает образование кист во взрослых почках, в которых отсутствуют различные предрасполагающие к кистам гены. 21,23 Мы предполагаем, что низкая скорость непрерывного обновления клеток во взрослой почке 31 является триггером для инициации образования кист клетками, дефицитными по Kif3a или Vhl / Kif3a , причем последние генотип более сенсибилизирован, чем первый, к инициации образования кисты. Мы не наблюдали увеличения скорости пролиферации мутантных клеток Vhl / Kif3a в ранний момент времени после делеции гена, утверждая, что повышенное инициирование образования кист не связано с изменением скорости клеточного обновления.Вероятным фактором, способствующим повышенному образованию кист, является нарушение регуляции плоскостного деления эпителиальных клеток. Во время развития или восстановления поврежденных почечных канальцев клеточные деления обычно ориентируются по длине канальца. Неправильно ориентированные клеточные деления в плоскости канальца увеличивают диаметр канальца, что является начальным этапом формирования кисты. 24 Первичная ресничка контролирует ориентированное клеточное деление посредством неканонических WNT и сигнальных путей планарной клеточной полярности; потеря ресничек или нескольких важных сигнальных компонентов ресничек вызывает дезориентацию клеточного деления в нормальных канальцах до образования кист. 24 pVHL также контролирует плоскость клеточного деления, регулируя стабильность астральной сети микротрубочек, тем самым обеспечивая правильную ориентацию митотического веретена. 32 Ишемическое повреждение, вызывающее клеточную пролиферацию в Vhl пустых почечных канальцах, увеличивало частоту неправильно ориентированных делений эпителиальных клеток, коррелируя с образованием микрокист у этих животных. 33 В соответствии с идеей, что VHL / MUTATION KIF3A / kif3a мутация компромисс два разных механизма, которые обеспечивают ориентированную ориемую клеточную дивизию, анафаз в цистическую эпителию Kif3a δ / δ ; VHL δ / Δ мыши были в четыре раза чаще дезориентированы из плоскости кистозного эпителия, чем у мышей Kif3a Δ / 4 9 мышейНаблюдение, что анафазы в KIF3A Δ 3 4 Δ 9 /
Делеция Vhl в эпителиальных клетках проксимальных канальцев мыши вызывала образование кист у небольшой части старых мышей. 34 Поскольку этот фенотип можно восстановить путем коделяции Arnt , кодирующего HIF1 β (партнер димеризации HIF1 α и HIF2 α , который необходим для активации транскрипции), но не путем коделяции Hif , 34 , возможно, что комбинированная активность HIF1 α и HIF2 α может способствовать образованию кист у мышей Kif3a Δ/Δ ; Vhl Δ/Δ .Недавно была предложена более общая роль стабилизации HIF1 α в развитии кисты. 19,20 Однако в этом исследовании мы обнаружили, что кистозные поражения, вызванные делецией Kif3a , не характеризуются стабилизацией HIF1 α , а делеция Hif1a не оказывает никакого влияния на образование кист в этой модели. . Стабилизация HIF1 α может быть ограничена более тяжелым поликистозом почек, чем достигается в этой модели, или изменения клеточной сигнализации, вызванные мутацией генов PKD1/PKD2 при аутосомно-доминантном поликистозе почек, могут способствовать активации HIF1 α в кистах через конститутивную мишень активации рапамицина у млекопитающих, 35–37 , которая вызывает усиленную трансляцию HIF1 α . 38,39
Наконец, эти исследования разработали и утвердили неинвазивный метод количественной оценки кистозной нагрузки у мышей, который должен оказаться полезным для будущих исследований, изучающих генетические основы и методы лечения поликистоза почек и рака почки.
Краткие методы
, 41 и/или Hif1a фл/фл 42 животных.Делеция гена у 6-недельных животных была достигнута путем кормления кормом с тамоксифеном (400 частей на миллион) от Harlan Laboratories в течение 2 недель. Для делеции гена у животных P2 кормящим самкам вводили внутрибрюшинно тамоксифен (0,1 мг/г массы тела) со дней P2–P4. BrdU (80 мкг г/г массы тела) вводили внутрибрюшинно за 18 часов до эвтаназии. Эксперименты на мышах проводились по экспериментальной лицензии 131/2012 Ветеринарного управления кантона Цюрих. Последовательности праймеров для генотипирования приведены в дополнительной таблице 1. мк КТ-визуализацияVisipaque 270 (GE Healthcare) вводили внутривенно в дозе 8 мкл л/г массы тела, как описано. 22 µ КТ-изображения были получены с помощью микроКТ Quantum FX (Perkin Elmer) со следующими настройками: 100 µ A, 90 кВ, стробирование дыхания и точное сканирование. µ Данные КТ были экспортированы в виде изображений DICOM для анализа вручную с помощью программного обеспечения Myrian (intrasense) для объемных измерений и трехмерной реконструкции, а также в формате данных Quantum FX для обзорных рентгеновских снимков.
Иммуногистохимия
Иммуногистохимию и иммунофлуоресценцию почек, фиксированных формалином и залитых парафином, проводили, как описано 43 , со следующими антителами: . Loffing), BrdU (MAP3510; EMD Millipore), расщепленная каспаза 3 (9661; Cell Signaling Technology), α -Glut1 (Ab14683; Abcam, Inc.), α -HIF1 α (NB100-105; Novus Biologicals), α -Ki67 (TEC-3; DakoCytomation), α -NaPi (подарок J.Biber), α -NCC (AB3553; EMD Millipore), α -THP (sc-20631; Santa Cruz Biotechnology) и α -ацетилированный тубулин (T6793; Sigma-Aldrich). Вторичные антитела, связанные с Alexa 488 или Alexa 568, были получены от Life Technologies. Вторичное антитело мыши α , связанное с пероксидазой хрена, было получено от Thermo Fisher Scientific.
Количественный ПЦР-анализ геномной ДНК
Фиксированные формалином и залитые в парафин срезы почек дважды промывали в ксилоле в течение 1 минуты и сушили на воздухе.Геномную ДНК выделяли из этих срезов или из измельченных в порошок целых почек с помощью набора для выделения ДНК Arcturus PicoPure (Life Technologies). ПЦР-анализ в реальном времени проводили с помощью SYBR FAST Universal 2× qPCR Master Mix, а праймеры перечислены в дополнительной таблице 1. гепарин. Креатинин и азот мочевины определяли с помощью клинической системы UniCel DxC 800 Synchron.
Благодарности
Мы благодарим Peter Igarashi за предоставление мышей, Nadine Nägele за помощь в измерениях плазмы, Thi Dan Linh Nguyen-Kim и Olivio Donati за помощь в количественном анализе изображений µ CT, а также Jürg Bieber, Carsten Wagner и Johannes Loffing для обеспечения антител.
Эта работа была поддержана Швейцарским национальным научным фондом (SNF Professorship PP00P3_128257) и Европейским исследовательским советом (стартовый грант 260316).
- Copyright © 2015 Американского общества нефрологов
стр.Мутации зародышевой линии N78S и p.R161Q гена VHL присутствуют при синдроме фон Хиппеля-Линдау в двух родословных
Введение
Болезнь Гиппеля-Линдау (ВГЛ) (OMIM 1) синдром аутосомно-доминантного семейного рака, предрасположенность к ряду доброкачественных и злокачественных опухолей, в том числе центральная нервная система (ЦНС) гемангиобластомы, почечные кисты и почечно-клеточный рак (ПКР), а также феохромоцитома (ПКК). Дополнительные опухоли, такие как опухоли поджелудочной железы и эндолимфатического мешка и цистаденомы придатка яичка и широкой связки, также были описано ранее (1–6).На синдром VHL приходится ~ 1/3 пациентов с гемангиобластомой ЦНС, > 50% пациентов с поражением сетчатки ангиома, 1% пациентов с ПКР, 50% пациентов с очевидно изолированный семейный РПЖ и 11% пациентов с очевидно спорадическим ПКК (7–9). ВГЛ присутствует на частоте 1 в 36 000 живорождений в Восточной Англии (2,5). Подобно другим наследственным синдромам, ВХЛ имеет свои клинические проявления. классификация на основе ассоциации генотип-фенотип. На основе клинические симптомы, пациенты с ПКР низкого риска (<5%) классифицируется как ВХЛ типа 1, а пациенты с РСС высокого риска (> 60%) — как ВХЛ тип 2.ВХЛ типа 2, по мнению 7-20% родов ВХЛ, является далее подразделяется на три гипотипа, 2А (пациенты с низким риск ПКР), 2B [пациенты с повышенным риском (~70%) ПКР] и 2C (пораженные члены с изолированным ПКР) (1,2,10).
VHL, ген, ответственный за болезнь VHL, был выявлен в 1993 г. (11). Это представляет собой плейотропный ген-супрессор опухоли, расположенный на хромосоме 3. (3p25.3), который широко экспрессируется в тканях плода и взрослого человека (11,12). Несколько зародышевых линий VHL мутации были идентифицированы у лиц с болезнью VHL, как а также соматические мутации в спорадических опухолях (5,13–16).Семьи VHL типа 1 часто содержат более крупные делеции и нонсенс. мутации в дополнение к миссенс-вариантам в гене VHL, а пациенты с болезнью 2 типа несут миссенс-мутации почти исключительно (5,10). До 96% пациентов с ПКР имеют миссенс-мутации (10).
Первый случай VHL с зародышевой мутацией в Китае было сообщено Huang et al (17) в 2004 г. В настоящем исследовании два частые мутации зародышевой линии VHL, c.A233G (p.N78S) и c.G482A (p.R161Q), описаны в двух семействах с типом 2A и 2С синдром ВХЛ.Пациенты были оперированы и впоследствии последовать.
Материалы и методы
пациентов
В этом исследовании были исследованы две родословные с синдромом VHL. учиться; семья 1, родословная из трех поколений, в том числе 6 особи (рис. 1А) и семьи 2, родословная из четырех поколений, содержащая 24 особи (рис. 1Б). Настоящее исследование было проведено в соответствии с Хельсинкской декларацией и был одобрен комитеты по этике 117-й больницы НОАК и провинции Чжэцзян Университет (Ханчжоу, Китай).Дано письменное информированное согласие всеми субъектами этого исследования.
В семье 1 пробанд (F1-II-1), 31 год. пациент мужского пола с жалобами на головную боль, пароксизмальную гипертензию (150–178/100–120 мм рт.ст.) и приступы головокружения в 2009 г. с 4-летней история. УЗИ (УЗИ) и магнитно-резонансная томография (МРТ) подтвержденные единичные массы, множественные опухоли и множественные кисты в правый надпочечник, двусторонняя почка и поджелудочная железа соответственно (Рис. 2А и Б). Следующий лечение α-адреноблокаторами, больному выполнена операция на правом надпочечнике опухолеэктомия и правая нефронсберегающая хирургия (NSS).Правый PCC и множественные ПКР справа (2 степень по Фурману) были подтверждены гистопатологическое исследование. В 2012 году пациенту был поставлен диагноз с двусторонними множественными узелками в головке придатка яичка. двусторонний Выполнена резекция придатка яичка и придатка яичка. папиллярная аденома подтверждена гистологическим исследованием. Дополнительно младший брат больного (F1-II-3, 30 лет) представлены множественными образованиями правой почки, узлами придатка яичка и множественные кисты поджелудочной железы (рис. 2С и Д). Произведена радикальная правосторонняя нефрэктомия и гистопатологический анализ показал правый ПКР с геморрагической кистой (Фурман 2 класс; табл. I).
Таблица IКлинические проявления ПКР и ПКР с мутациями VHL. |
Таблица I
Клинические проявления ПКР и ПКР с мутациями VHL.
| Индивидуум | Пол | Возраст на момент начала заболевания (лет) | ВХЛ мутация | Катехоламиновая сыворотка уровень (нг/л) (норма: DA, 100; E, 600; NE, 100) | Размер опухоли (см) | Операция | Гистология | |
|---|---|---|---|---|---|---|---|---|
| PCC | RCC | |||||||
| F1-II-1 | M | 31 | 66 с.N78S | — | R, 4,1×3,4×3,9 | R, 1,0×1,0×1,0; л, 1,0×1,0×0,8 | Р-Опухольэктомия; Р-НСС; Запланированные левые NSS позже | R-PCCC, BI-RCC |
| F1-II-3 | M | 30 | P.N78S | — | — | R, 4,0 × 3.0 × 2.5 | Радикальный R-nephrectomy | R-RCC |
| F1-III-1 | M | 5 | P.N78S | — | — | — | — | — |
| F2-II -11 | Ж | 18 | р.R161Q | — | R, 5.5 × 4.5 × 2.8 | — | Tumorectome | R-PCCC |
| F2-III-9 | M | 9 (R) / 17 (L) | P .R161Q | Приподнятый (DA, 88,64; Е, 106,23; NE, 827,15)/повышенный (DA, 600,36; E, 297,94; NE, 1093,84) | л, 7,0×5,5×3,4; Р, 5.6 × 3.8 × 3.0 | — | Tumorectomy | BI-PCC / HE |
| F2-III-4 | F | 28 | P.R161Q | Rived (DA, 268.63; Е 1270,3; СВ, 1802,25) | Д, 6,0×4,7×3,5; Р, 3,5×2,8×2,5 | — | Двучастное адреналэктомия | Bi-PCC/Si |
В семье 2 пробанд (F2-II-11), 45 лет. пациентка с пароксизмальной гипертензией (180–210/100–120 мм рт. ст.), головная боль, сердцебиение и гипергидроз в 1985 г., в возрасте 18 лет. УЗИ показало изолированное новообразование в правый надпочечник. После лечения альфа-адреноблокаторами и феноксибензамина или ортеразозина гидрохлорид, пациенту было проведено опухолеэктомия правого надпочечника, и РСС был подтвержден гистопатологическое исследование.В 2002 году сын пробанда (F2-III-9, 9 лет) с пароксизмальной гипертензией. (140–160/100–110 мм рт. ст.) и головная боль. Биохимическое исследование указывают на повышение уровня катехоламинов в сыворотке и изолированное образование в правый надпочечник подтвержден УЗИ и КТ. Следующий лечение α-адреноблокаторами, выполнена опухолеэктомия правого надпочечника. Гистопатологический анализ выявил ПКР правого надпочечника. В 2010, в возрасте 17 лет лапароскопическая резекция опухоли надпочечников с углекислотным пневмоперитонеумом на основании признаки пароксизмальной гипертензии, головная боль, повышение уровня сыворотки катехоламины и образование в левом надпочечнике, но без аномалии правого надпочечника.ПКК был подтвержден гистопатологическое исследование. Другая пациентка, племянница пробанд (F2-III-4, 33 года), поступил с жалобами на головную боль, устойчивая гипертензия, приступы головокружения и избыток сыворотки катехоламины. Двусторонние образования в надпочечниках были подтвержденный УЗИ и КТ, когда пациенту было 28 лет в 2007. Блокада α1 в высоких дозах мезилатом доксазозина и Гидрокортизон кортизол вводили перед операцией и частичная двусторонняя лапароскопическая адреналэктомия с углекислым газом наложен пневмоперитонеум.Гистопатологическое исследование указан двусторонний ПКР. 38-летний отец этого пациента (F2-II-3, брат пробанда) скончался от гипертонической внутримозговое кровоизлияние за 21 год до этого исследования (таблица I). Среди членов семьи 2, у троих был диагностирован РПЖ как единственный симптом и никаких других систематические опухоли, в том числе гемангиобластомы ЦНС, почечные кисты или ПКР были выявлены.
Все пять пациентов (F1-II-1, F1-II-3, F2-II-11, F2-III-4 и F2-III-9).Биохимия и визуализация обследование не выявило признаков рецидива ПКР и ПКР и АД нормализовались, за исключением один случай (пациент F1-II-1), где левый ПКР оставался очевидным, а пациент согласился на плановую левую NSS на более позднем этапе. Пациент F2-III-4 нуждался в заместительной терапии стероидами. Тем не мение, пациенты F2-III-4 и F2-III-9 с двусторонней опухолеэктомией или парциальная адреналэктомия не привела к развитию адренокортикальной недостаточности или аддисоновский криз в период наблюдения.
Анализ мутаций
Образцы периферической крови были взяты на мутацию анализ протоонкогена RET и опухоли VHL ген супрессор. Геномная ДНК была извлечена из образцов цельной крови. с помощью набора крови QIAamp (Qiagen, Hilden, Германия) согласно инструкции производителя. Полимеразная цепная реакция (ПЦР) амплификация всех экзонов и фланкирующих сплайс-соединений Выполняли РЭТ и ВХЛ с последующим прямым двунаправленное секвенирование ДНК. Сто несвязанных элементов управления с аналогичное этническое происхождение также было включено в это исследование.
Результаты
В семье 1 с синдромом ВХЛ 2А типа выявлены гетерозиготный вариант, c.A233G (p.N78S), в экзоне 1 Ген VHL в двух пораженных случаях и одном клинически нормальном член семьи (F1-III-1, 5-летний сын пробанда; рис. 3А). Гетерозиготный нуклеотид замена в экзоне 3 гена VHL, c.G482A (p.R161Q), идентифицирован у пробанда (F2-II-11) и двух дополнительные случаи (F2-III-4 и F2-III-9) семейства 2 с типом VHL 2С (фиг. 3В). Никаких мутантов В этих случаях был идентифицирован ген RET.
Обсуждение
ПКР, связанный с заболеванием VHL, представляет собой опухоль симпатический параганглий, мозговое вещество надпочечников, которое осложнено по гипертонии. PCC обычно доброкачественный, с ~ 20% злокачественных случаев. (18). Молекулярно-генетические исследования предполагают, что ~ 50% людей с явной семейной несиндромальные РПЖ имеют мутации зародышевой линии VHL (18). Предыдущие исследования показали, что ПКК возникает спорадически или как часть наследственной предрасположенности к опухолям синдромы, вызванные мутациями зародышевой линии, поражающими один из десяти следующие гены предрасположенности: VHL, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2, NF1, TMEM127 или MAX (таблица II) (1,2,19–27).Большинство семейных случаев ПКР и 10–20% спорадических случаев несут зародышевые мутации этих генов (4). ПКР составляет 90–95% новообразований. возникающие из почек, что является важной причиной смерти в Болезнь ВХЛ (7). ПКР встречается редко детей и молодых взрослых, и происходит как в несемейном, так и в семейная форма. Наследственный ПКР составляет ~ 4% ПКР, в том числе следующие расстройства: болезнь ВХЛ; наследственный лейомиоматоз связанный с мутацией гена FH; туберозный склероз, который возникает в результате мутации зародышевой линии TSC1 и TSC2 гены; наследственный папиллярный почечно-клеточный рак (HPRCC), который связанный с геном МЕТ; и ген, ответственный за Birt Синдром Хогга-Дюбе, который, как сообщается, является FLCN. ген (табл. II) (28–31).При семейной форме ПКР возникает в раннем возрасте (за исключением HPRCC) и обычно проявляется двусторонним поражением с высокой частотой (7,31). Согласно недавнему исследованию, 20/255 больных (7,8%) с наследственным лейомиоматозом и ПКР. сообщалось о первичном поражении надпочечников, в том числе у 4 пациентов с двустороннее поражение надпочечников и 4 с множественными узелками (28). Таким образом, в будущих исследованиях PCC и пациентов с ПКР, целевые регионы (все экзоны VHL, РЕТ, SDHA, SDHB, SDHC, SDHD, SDHAF2, NF1, TMEM127, МАКС, ФХ, TSC1, TSC2, MET и FLCN) или экзом следует проанализировать последовательность (32).
Таблица IIРезюме генов восприимчивости связаны с ПКР и ПКР. |
Таблица II
Резюме генов восприимчивости связаны с ПКР и ПКР.
| Джин (местонахождение) | Белок функция | Клиническая фенотип | (Ref.) |
|---|---|---|---|
| RET (10q11.2) | Тирозинкиназа рецептор, играющий ключевую роль в регуляции клеточного пролиферация, миграция, дифференцировка и выживание посредством эмбриогенез. | Множественные эндокринные неоплазия 2 типа (МЭН 2) – наследуется по аутосомно-доминантному типу предрасположенность к медуллярному раку щитовидной железы и ПКР. МУЖЧИНЫ 2А характеризуется медуллярным раком щитовидной железы, ПКР и первичным гиперпаратиреоз. МЭН 2В характеризуется медуллярным поражением щитовидной железы. карцинома, ПКР и аномалии развития (например, марфаноидный габитус и невромы слизистых оболочек). КПК при МЭН 2 характерны для надпочечников. и доброкачественный. | (22) |
| ВХЛ (3p25) | Короткий белок с мощная опухолесупрессивная активность. | Фон Хиппель-Линдау заболевание, аутосомно-доминантная предрасположенность к ПКР, гемангиобластомы сетчатки и мозжечка, чистый ПКР, несекреторный нейроэндокринные опухоли поджелудочной железы, эндолимфатические опухоли и висцеральные кисты (почечные, поджелудочные и эпидидимальные). Редко голова и шея параганглиомы. Эпидидимальные цистаденомы. | (1) |
| SDHA (5p15) | Флавопротеин каталитическая субъединица Комплекса II. | Аутосомный доминантно наследуемая предрасположенность к ПКР головы и шеи параганглиомы (хотя и нечастая причина этих опухолей) (аутосомно рецессивно наследуемая ювенильная энцефалопатия или неонатальная кардиомиопатия). | (23) |
| СДХБ (1p36) | Железо-серный белок каталитическая субъединица Комплекса II. | Аутосомный доминантно наследуемая предрасположенность к ПКР головы и шеи параганглиомы и почечно-клеточный рак. Высокая частота вненадпочечниковый РСС и злокачественные новообразования. | (24) |
| SDHC (1q21) | Один из двух трансмембранные субъединицы Комплекса II дыхательной цепи. | Аутосомный доминантно наследуемая предрасположенность головы и шеи параганглиомы, ПКР. | (19) |
| SDHD (11q23) | Один из двух трансмембранные субъединицы Комплекса II дыхательной цепи. | Аутосомный доминантно наследуемая предрасположенность к парасимпатическим головным и параганглиомы шеи и ПКР. Развитие опухоли обычно происходит только у лиц, унаследовавших мутацию от своего отец (т. е. влияние родителя на фенотип заболевания). | (20) |
| SDHAF2 (11к12.2) | Митохондриальный фактор сборки для Комплекса II, взаимодействует непосредственно с SDHA. | Аутосомный доминантно наследуемая предрасположенность к параганглиомам головы и шеи и ПКК. | (25) |
| NF1 (17q11.2) | Исследования на животных модели показывают, что гомозиготные мутантные мыши по гену NF1 проявляют различные аномалии развития, ведущие к среднему сроку беременности летальность. | Нейрофиброматоз Тип 1 (болезнь фон Реклингхаузена) наследуется по аутосомно-доминантному типу предрасположенность к ПКР, опухоли периферической нервной системы (кожной, узловые и плексиформные нейрофибромы), кишечные и гастроинтестинальные стромально-клеточные опухоли, злокачественные глиомы и ювенильные хромовые лейкозы. | (26) |
| ТМЕМ127 (2q11.2) | Трансмембранный белок, участвующий в транспортировке белков. | Аутосомный доминантно наследуемая предрасположенность к ПКР и головной и шейной параганглиомы. Относительно поздний средний возраст постановки диагноза (по сравнению с Болезнь VHL и мутации SDHB). | (27) |
| МАКС (14q23) | Компонент сети MYC-MAX-MXD1 и регулирует пролиферацию клеток, дифференцировка и апоптоз. | Аутосомно-доминантный наследование двустороннего ПКР, подтверждающее связь между генетические аномалии и множественные опухоли. Ассоциация с также было обнаружено злокачественное новообразование. | (21) |
| FH (1q42–43) | FH — цикл Кребса фермент, превращающий фумарат в малат. | Наследственный лейомиоматоз и ПКР — аутосомно-доминантное заболевание характерна семейная с агрессивной формой папиллярной почки рак и развитие кожных и маточных лейомиомы. | (28) |
| TSC1 (9q34) и TSC2 (16p13.3) | Белки действуют как супрессоры опухолевого роста, агенты, регулирующие пролиферацию клеток и дифференциация. | Туберозный склероз или комплекс туберозного склероза является редким мультисистемным генетическим заболеванием. заболевание, вызывающее рост доброкачественных опухолей в головном мозге и на другие жизненно важные органы, такие как почки, сердце, глаза, легкие и кожа. | (29) |
| МЕТ (7q31) | MET трансмембранный белок представляет собой рецептор тирозинкиназы, который индуцирует пролиферацию и дифференцировка эпителиальных и эндотелиальных клеток, клеточных ветвление и инвазия. | Наследственный папиллярная карцинома почки – аутосомно-доминантный рак почки синдром, при котором пострадавшие люди подвержены риску развития многоочаговый двусторонний папиллярный рак почки 1 типа. | (30) |
| FLCN (17q11.2) | FLCN формирует комплекс с фолликулин-взаимодействующими белками (FNIP1 и FNIP2). Эти компоненты взаимодействуют с АМФ-активируемой протеинкиназой. | Бирт-Хогг-Дюбе синдром является аутосомно-доминантным заболеванием, и больные люди развиваются кожные фиброфолликуломы, легочные кисты, спонтанные пневмоторакс и опухоли почек. | (31) |
Ген VHL консервативен на протяжении эволюции и встречается у всех многоклеточных организмов. Две ВХЛ продукты гена, pVHL, полноразмерный белок 30 кДа [p30, 213 амино кислоты, NM_000551.2 (вариант 1 мРНК)] и белок массой 19 кДа [p19, 160 аминокислот, NM_198156.1 (вариант 2 мРНК)], подавляет опухоль у голых мышей и регулируют индуцируемый гипоксией фактор-α (HIF-α) (33,34). pVHL имеет два домена, домен β (N-концевой домен β-листа), который содержит макромолекулярный сайт связывания и α-домен (N-концевой α-спиральный домен), который взаимодействует с элонгином С (35).Область между кодонами 75–82 расположена в β-связывающем домене. белка VHL; таким образом, мутация p.N78S мешает HIF связывание, что приводит к чрезмерной транскрипции мишени HIF гены, такие как фактор роста эндотелия сосудов (13). Кодон 161 расположен в α-домене. и взаимодействует с элонгином С. Мутация в домене α затрагивает Опухолесупрессорная активность VHL (36,37). Из-за ошибок в хромосомной репликации во время клеточного деления дополнительные гены или модифицирующие факторы могут быть вовлечены в генетическая основа VHL (37).
корреляции генотип-фенотип VHL были ранее установленный (5,17). Мутация p.N78S была сообщалось, что он связан с ПКР, ПКР и гемангиобластомами ЦНС (8). Мутация p.R161Q была впервые описан Tong et al (38) у китайских пациентов с VHL, перенесших несиндромальный семейный ПКР без артериальной гипертензии, также известный как быть связанным с гемангиобластомой сетчатки и ЦНС и ПКР (10). Для пациентов, включенных в В настоящем исследовании мутации VHL соответствовали фенотипы типа 2А и 2С.В частности, p.R161Q был ассоциирован с ПКР с гипертензионным синдромом, а p.N78S имел более тяжелое течение клинический фенотип (ПКР и двусторонний ПКР) по сравнению с p.R161Q. Однако ни одна из мутаций не была связана с ЦНС. гемангиобластомы.
Идентификация опухоли ВХЛ ген-супрессор облегчает диагностику болезни VHL, особенно у пациентов, не отвечающих клиническим диагностическим критерии. Это также имеет важное значение для оценки риск развития дальнейших опухолей у пациентов и ВХЛ заболевания у своих родственников.В семье 1 молодой член семьи (F1-III-1, возраст 5 лет) не имели выраженного клинического фенотипа. но нес мутацию p.N78S. Идентификация Ген-супрессор опухоли VHL полезен для раннего профилактика и лечение синдрома ВХЛ. Кроме того, среднее возраст на момент постановки диагноза ПКР и ПКР при ВХЛ составляет ~30 и ~40 лет, соответственно (39,40). Однако, по данным клинического отчет, у 2,75-летней пациентки с ПКР было обнаружено, что она является носителем мутация зародышевой линии p.R161Q (40). На сегодняшний день это самый молодой пациент с ПКР.За пациентов в настоящем исследовании, возраст начала для пациента F2-III-9 было 9 лет, а двум другим пациентам было 18 и 28 лет. лет, когда им поставили диагноз РСС. Таким образом, молекулярный генетическое тестирование и генетическое консультирование пациентов с РПЖ и ПКР следует проводить в раннем возрасте. Ранняя диагностика ВХЛ было бы полезно, так как регулярное наблюдение за опухолями и раннее вмешательство рекомендуется для улучшения прогноза.
Многочисленные исследования показали, что пациенты с ВХЛ при лечении НСС высока частота местных рецидивов новых первичные опухоли, но низкий риск отдаленных метастазов.Примерно У 25% пациентов с ВХЛ с более распространенным ПКР (>3 см) развивается метастатическое заболевание (41,42). Радикальная двусторонняя нефрэктомия должна следует проводить при ПКР >3 см и трансплантации почки. обязательный. Двусторонний ПКР обычно лечится CSA (надпочечниковая туморэктомия или частичная адреналэктомия). Пациенты могут избегать приема гормонов замена, но будет риск позднего рецидива. Всего адреналэктомия может рассматриваться как альтернатива при наличии высокая частота множественных двусторонних образований надпочечников и больших опухоли; однако требуется пожизненная замена стероидов.Все пациенты должны регулярно наблюдаться для выявления процессирующей адренокортикальной недостаточности или аддисонова криза (3).
В настоящем исследовании пациент с мутацией p.N78S (F1-II-1) с множественным двусторонним ПКР (1,0 и 1,0 см) и правым PCC (4,1 см) подвергли правой NSS и правой CSA. Слева ПКР наблюдали с помощью визуализации и не проявляли выраженного увеличение, в то время как локорегиональный рецидив, отдаленные метастазы и/или ЦНС не были обнаружены в двух случаях (F1-II-1 и F1-II-3) 39 и через 6 мес после операции соответственно.Среди трех p.R161Q мутации, у одного (F2-III-9) был левый ПКР через 8 лет после поступил с правым ПКР в возрасте 9 лет, у одного (F2-III-4) одновременный двусторонний ЗПК и другой (F2-II-11) представлены только с правым ПКР в 18 лет. Все они прошли двусторонний или односторонний ЦСА и функция коры надпочечников в норме. без замены стероидов. Таким образом, идентификация основная мутация не только подтверждает диагноз, но и предоставляет полезную информацию для генетического консультирования и последующего принятие управленческих решений.
Благодарности
Авторы благодарят всех пациентов и их семьи, которые согласились участвовать в этом исследовании. Это исследование было при поддержке Ключевого научно-исследовательского проекта Нанкина Военное командование, Китай (09Z038 и 10Z036) и Национальный 973 Программа фундаментальных исследований Китая (2013CB0)
Каталожные номера
1 | Хуан И, Чжоу Д, Лю Дж, Чжоу П, Ли С и Ван Зи: зародышевые мутации гена VHL у семи китайцев семьи с болезнью Гиппеля-Линдау.Int J Mol Med. 29:47–52. 2012. PubMed/NCBI . |
2 | Qi XP, Ma JM, Du ZF и др.: зародышевая линия RET мутации, идентифицированные секвенированием экзома у китайского множественного эндокринная неоплазия типа 2А/семейная медуллярная карцинома щитовидной железы семья. ПЛОС Один. 6:e203532011. Просмотр статьи : Академия Google |
3 | Qi XP, Chen XL, Ma JM и др.: RET протоонкогенный генетический скрининг семей с множественными эндокринная неоплазия 2 типа оптимизирует диагностические и клинические управления в Китае.Щитовидная железа. 22:1257–1265. 2012. Просмотр статьи : Google Scholar |
4 | Хименес-Рокепло AP, Дахия PL и Робледо М: Последние сведения о генетике параганглиомы, феохромоцитомы, и сопутствующие наследственные синдромы. Горм Метаб Рез. 44:328–333. 2012. Просмотр статьи: Google Scholar: PubMed/NCBI . |
5 | Вича А., Хользерова М., Крепелова А. и др.: Молекулярно-цитогенетическая характеристика у четырех детей феохромоцитомы и параганглиомы.Патол Онкол Рез. 17:801–808. 2011. Просмотр статьи: Google Scholar: PubMed/NCBI . |
6 | Сантарпия Л., Лапа Д. и Бенвенга С.: Мутация зародышевой линии гена фон Хиппеля-Линдау (VHL) 695 G>A (R161Q) у пациента со своеобразным фенотипом с ВХЛ 2С типа синдром. Энн Н.Ю. Академия наук. 1073: 198–202. 2006. Просмотр статьи: Google Scholar: PubMed/NCBI . |
7 | Ричардс Ф.М., Пейн С.Дж., Збар Б., Аффара Н.А., Фергюсон-Смит М.А. и Махер Э.Р.: Молекулярный анализ de novo зародышевые мутации в гене болезни фон Гиппеля-Линдау.Хум Мол Жене. 4:2139–2143. 1995. Просмотр статьи: Google Scholar: PubMed/NCBI . |
8 | Neumann HP, Bausch B, McWhinney SR и др.: Мутации зародышевой линии при несиндромальной феохромоцитоме. N Engl J Med. 346: 1459–1466. 2002. Просмотр статьи: Google Scholar: PubMed/NCBI . |
9 | Neumann HP, Bender BU, Berger DP и др.: Распространенность, морфология и биология почечно-клеточного рака у фон Болезнь Гиппеля-Линдау по сравнению со спорадической почечно-клеточной карциномой.Дж Урол. 160: 1248–1254. 1998. Просмотр статьи: Google Scholar: PubMed/NCBI . |
10 | Гонг К., Чжан Н., Чжан К. и На И.: связь сверхэкспрессии эритропоэтина с фон Мутации гена опухолевого супрессора Хиппеля-Линдау между индуцируемый гипоксией фактор-1α и -2α в спорадических светлоклеточных почечных карцинома. Int J Mol Med. 26:907–912. 2010. PubMed/NCBI . |
11 | Латиф Ф., Тори К., Гнарра Дж. и др.: Идентификация опухолевого супрессора болезни фон Гиппеля-Линдау ген.Наука. 260:1317–1320. 1993. Просмотр статьи: Google Scholar: PubMed/NCBI . |
12 | Кэнтли Дж., Селман С., Шукла Д. и др.: Делеция гена фон Хиппеля-Линдау в бета-клетках поджелудочной железы нарушает гомеостаз глюкозы у мышей. Джей Клин Инвест. 119:125–135. 2009. PubMed/NCBI . |
13 | Кросси П.А., Ричардс Ф.М., Фостер К. и др.: Выявление внутригенных мутаций в гене фон Хиппеля-Линдау. ген-супрессор опухоли и корреляция с заболеванием фенотип.Хум Мол Жене. 3: 1303–1308. 1994. Просмотр статьи: Google Scholar: PubMed/NCBI . |
14 | Главац Д., Нейманн Х.П., Виттке С. и др.: Мутации в гене супрессора опухоли VHL и связанные с ними поражения в семьях с болезнью фон Гиппеля-Линдау из Центральной Европы. Гул Жене. 99: 271–280. 1996. PubMed/NCBI . |
15 | Энг С., Кросси П.А., Маллиган Л.М. и др.: Мутации в протоонкогене RET и фон Хиппеля-Линдау ген-супрессор опухоли при спорадических и синдромальных феохромоцитомы.J Med Genet. 32:934–937. 1995. Просмотр статьи: Google Scholar: PubMed/NCBI . |
16 | Кондо К., Яо М., Йошида М. и др.: Комплексный мутационный анализ гена VHL при спорадических почечных клеточная карцинома: связь с клинико-патологическими параметрами. Гены Хромосомы Рак. 34:58–68. 2002. Просмотр статьи: Google Scholar: PubMed/NCBI . |
17 | Huang YR, Zhang J, Wang JD и Fan XD: Генетическое исследование большого китайского родства с фон Хиппель-Линдау болезнь.Чин Мед Дж (англ.). 117: 552–557. 2004. PubMed/NCBI . |
18 | Опочер Г. и Скьяви Ф.: Генетика феохромоцитомы и параганглиомы. Best Pract Res Clin Endocrinol Метаб. 24:943–956. 2010. Просмотр статьи: Google Scholar: PubMed/NCBI . |
19 | Печковска М., Каскон А., Прейбиш А. и др.: Вненадпочечниковые и надпочечниковые феохромоцитомы, связанные с зародышевая мутация SDHC.Nat Clin Pract Endocrinol Metab. 4:111–115. 2008. Просмотр статьи: Google Scholar: PubMed/NCBI . |
20 | Астути Д., Дуглас Ф., Леннард Т.В. и др.: Мутация зародышевой линии SDHD при семейной феохромоцитоме. Ланцет. 357: 1181–1182. 2001. Просмотр статьи: Google Scholar: PubMed/NCBI . |
21 | Комино-Мендес I, Грасиа-Азнарес Ф.Х., Schiavi F, et al: Секвенирование экзома идентифицирует мутации MAX как Причина наследственной феохромоцитомы.Нат Жене. 43:663–667. 2011. Просмотр статьи : Академия Google : PubMed/NCBI |
22 | Энг С, Клейтон Д, Шуффенекерет I и др. al: Взаимосвязь между специфическими мутациями протоонкогена RET и фенотип заболевания при множественной эндокринной неоплазии 2 типа. Анализ международного консорциума мутаций RET. ДЖАМА. 276: 1575–1579. 1996. Просмотр статьи : Google Scholar |
23 | Корпершук Э., Фавье Дж., Гаал Дж. и др.: Иммуногистохимия SDHA выявляет мутации гена SDHA зародышевой линии у по-видимому, спорадические параганглиомы и феохромоцитомы.Джей Клин Эндокринол Метаб. 96: E1472–E1476. 2011. Просмотр статьи: Google Scholar: PubMed/NCBI . |
24 | Астути Д., Латиф Ф., Даллол А. и др.: Джин мутации в субъединице сукцинатдегидрогеназы SDHB вызывают предрасположенность к семейной феохромоцитоме и семейной параганглиома. Am J Hum Genet. 69:49–54. 2001. Просмотр статьи: Google Scholar |
25 | Bayley JP, Kunst HP, Cascon A и др.: Мутации SDHAF2 при семейных и спорадических параганглиомах и феохромоцитома.Ланцет Онкол. 11: 366–372. 2010. Просмотр статьи: Google Scholar: PubMed/NCBI . |
26 | Бауш Б., Кошкер А.С., Фасснахт М. и др.: Комплексное сканирование мутаций NF1 в очевидно спорадических случаях феохромоцитомы. J Clin Endocrinol Metab. 91:3478–3481. 2006. Просмотр статьи : Академия Google : PubMed/NCBI |
27 | Нойманн Х.П., Салливан М., Винтер А. и др.: Мутации зародышевой линии гена TMEM127 у пациентов с параганглиомы головы и шеи и вненадпочечниковой локализации брюшной полости.Дж Клин Эндокринол Метаб. 96: E1279–E1282. 2011. Просмотр статьи: Google Scholar: PubMed/NCBI . |
28 | Shuch B, Ricketts CJ, Vocke CD и др.: Узловая гиперплазия надпочечников при наследственном лейомиоматозе и почечной недостаточности. клеточный рак. Дж Урол. 189: 430–435. 2013. Просмотр статьи: Google Scholar: PubMed/NCBI . |
29 | Дабора С.Л., Йозвяк С., Франц Д.Н. и др.: Мутационный анализ в группе из 224 больных туберозным склерозом указывает на повышенную тяжесть TSC2 по сравнению с TSC1, заболевание во многих органах.Am J Hum Genet. 68:64–80. 2001. Просмотр статьи: Google Scholar: PubMed/NCBI . |
30 | Шмидт Л., Юнкер К., Вейрих Г. и др.: Два Североамериканские семьи с наследственной папиллярной карциномой почки и идентичные новые мутации в протоонкогене МЕТ. Рак Рез. 58: 1719–1722. 1998. PubMed/NCBI . |
31 | Торо Дж. Р., Вэй М. Х., Гленн Г. М. и др.: BHD мутации, клинические и молекулярно-генетические исследования Синдром Берта-Хогга-Дюбе: новая серия из 50 семей и обзор опубликованных отчетов.J Med Genet. 45:321–331. 2008. PubMed/NCBI . |
32 | Qi XP, Du ZF, Ma JM и др.: Genetic диагностика аутосомно-доминантного поликистоза почек целенаправленный захват и секвенирование следующего поколения: полезность и ограничения. Ген. 516:93–100. 2013. Просмотр статьи: Google Scholar: PubMed/NCBI . |
33 | Илиопулуос О., Леви А.П., Цзян С., Кэлин В.Г. Младший и Голдберг М.А.: Негативная регуляция генов, индуцируемых гипоксией белком фон Хиппеля-Линдау.Proc Natl Acad Sci USA. 93:10595–10599. 1996. Просмотр статьи: Google Scholar: PubMed/NCBI . |
34 | Стеббинс К.Э., Келин В.Г. младший и Павлетич NP: Структура комплекса VHL-ElonginC-ElonginB: последствия для опухолесупрессорной функции VHL. Наука. 284: 455–461. 1999. Просмотр статьи : Академия Google : PubMed/NCBI |
35 | Окуда Х., Хираи С., Такаки Ю. и др.: Прямой взаимодействие бета-домена белка-онкосупрессора VHL с регуляторный домен атипичных изотипов ПКС.Биохим Биофиз Рез коммун. 263: 491–497. 1999. Просмотр статьи: Google Scholar: PubMed/NCBI . |
36 | Domene C и Illingworth CJ: Эффекты точечные мутации в pVHL при связывании HIF-1α. Белки. 80:733–746. 2012. |
37 | Арджуманд В. и Султана С.: Роль гена VHL Мутация при почечно-клеточном раке человека. Опухоль биол. 33:9–16. 2012. Просмотр статьи : Академия Google : PubMed/NCBI |
38 | Тонг А.Л., Цзэн З.П., Ли Х.З. и др.: фон Мутация гена Хиппеля-Линдау при несиндромальном семейном феохромоцитомы.Энн Н.Ю. Академия наук. 1073: 203–207. 2006. Просмотр статьи: Google Scholar: PubMed/NCBI . |
39 | Акчаглар С., Явашкаоглу И., Вурускан Х. и Октай Б: Генетическая оценка болезни Гиппеля-Линдау на ранних стадиях диагностика и улучшение прогноза. Инт Урол Нефрол. 40:615–620. 2008. Просмотр статьи: Google Scholar: PubMed/NCBI . |
40 | Совинз П. |